Büchner–Curtius–Schlotterbeck reaction
The Buchner–Curtius–Schlotterbeck reaction is the reaction of aldehydes or ketones with aliphatic diazoalkanes to form homologated ketones.[1] It was first described by Eduard Buchner and Theodor Curtius in 1885[2] and later by Fritz Schlotterbeck in 1907.[3] Two German chemists also preceded Schlotterbeck in discovery of the reaction, Hans von Pechmann in 1895 and Viktor Meyer in 1905.[4][5] The reaction has since been extended to the synthesis of β-keto esters from the condensation between aldehydes and diazo esters.[6] The general reaction scheme is as follows:

The reaction yields two possible carbonyl compounds (I and II) along with an epoxide (III). The ratio of the products is determined by the reactant used and the reaction conditions.
Reaction Mechanism
The general mechanism is shown below. The resonating arrow (1) shows a resonance contributor of the diazo compound with a lone pair of electrons on the carbon adjacent to the nitrogen. The diazo compound then does a nucleophilic attack on the carbonyl-containing compound (nucleophilic addition), producing a tetrahedral intermediate (2). This intermediate decomposes by the evolution of nitrogen gas forming the tertiary carbocation intermediate (3).

The reaction is then completed either by the reformation of the carbonyl through an 1,2-rearrangement or by the formation of the epoxide. There are two possible carbonyl products: one formed by migration of R1 (4) and the other by migration of R2 (5). The relative yield of each possible carbonyl is determined by the migratory preferences of the R-groups.
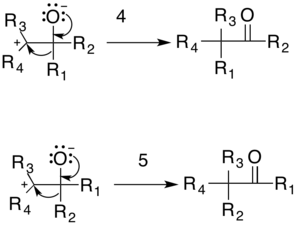
The epoxide product is formed by an intramolecular addition reaction in which a lone pair from the oxygen attacks the carbocation (6).

This reaction is exothermic due to the stability of nitrogen gas and the carbonyl containing compounds. This specific mechanism is supported by several observations. First, kinetic studies of reactions between diazomethane and various ketones have shown that the overall reaction follows second order kinetics.[7] Additionally, the reactivity of two series of ketones are in the orders Cl3CCOCH3 > CH3COCH3 > C6H5COCH3 and cyclohexanone > cyclopentanone > cycloheptanone > cyclooctanone.[7][8] These orders of reactivity are the same as those observed for reactions that are well established as proceeding through nucleophilic attack on a carbonyl group.
Scope and Variation
The reaction was originally carried out in diethyl ether and routinely generated high yields due to the inherent irreversibly of the reaction caused by the formation of nitrogen gas. Though these reactions can be carried out at room temperature, the rate does increase at higher temperatures. Typically, the reaction is carried out at less than refluxing temperatures.[3] The optimal reaction temperature is determined by the specific diazoalkane used. Reactions involving diazomethanes with alkyl or aryl substituents are exothermic at or below room temperature.[9] Reactions involving diazomethanes with acyl or aroyl substituents require higher temperatures.[9] The reaction has since been modified to proceed in the presence of Lewis acids and common organic solvents such as THF and dichloromethane. Reactions generally run at room temperature for about an hour, and the yield ranges from 70%-80% based on the choice of Lewis acid and solvent.[10]
Steric Effects
Steric effects of the alkyl substituents on the carbonyl reactant have been shown to affect both the rates and yields of Büchner–Curtius–Schlotterbeck reaction. Table 1 shows the percent yield of the ketone and epoxide products as well as the relative rates of reaction for the reactions between several methyl alkyl ketones and diazomethane.[7]
| Starting Ketone | Ketone % | Epoxide % | Relative Rate |
|---|---|---|---|
| CH3COCH3 | 38 | 33.5 | 1.0 |
| CH3COCH2CH3 | 32 | 40 | 0.4 |
| CH3COCH2CH2CH3 | 18 | 55 | 0.15 |
| CH3COCH(CH3)CH2 | - | - | 0.095 |
| CH3CO(CH2)8CH3 | 0 | 100 | - |
The observed decrease in rate and increase in epoxide yield as the size of the alkyl group becomes larger indicates a steric effect.
Electronic Effects
Ketones and aldehydes with electron-withdrawing substituents react more readily with diazoalkanes than those bearing electron-donating substituents (Table 2). In addition to accelerating the reaction, electron-withdrawing substituents typically increase the amount of epoxide produced (Table 2).
| Starting Compound | Carbonyl % | Epoxide % | Reference |
|---|---|---|---|
| Benzaldehyde | 97 | - | [3] |
| o-Nitrobenzaldehyde | 16.5 | 65 | [11] |
| p-Nitrobenzaldehyde | 29 | 46 | [11] |
| Piperonal | 31-46 | 18 | [12] |
| Acetone | 20-38 | 33-40 | [13][14] |
| Chloroacetone | Some | 65 | [11][13] |
| Trichloroacetone | Trace | 90 | [11][13] |
| Methoxyacetone | - | 39 | [8] |
| Cyclohexanone | 65 | 15 | [15] |
| 2-Chlorocyclohexanone | 11 | 50 | [16] |
| 2-Hydroxycyclohexanone | - | 90 | [17] |
The effects of substituents on the diazoalkanes is reversed relative to the carbonyl reactants: electron-withdrawing substituents decrease the rate of reaction while electron-donating substituents accelerate it. For example, diazomethane is significantly more reactive than ethyl diazoacetate, though less reactive than its higher alkyl homologs (e.g. diazoethane).[18][19][20][21][22][23] Reaction conditions may also affect the yields of carbonyl product and epoxide product. In the reactions of o-nitrobenzaldehyde,[11] p-nitrobenzaldehyde,[24] and phenylacetaldehyde[7] with diazomethane, the ratio of epoxide to carbonyl is increased by the inclusion of methanol in the reaction mixture. The opposite influence has also been observed in the reaction of piperonal with diazomethane, which exhibits increased carbonyl yield in the presence of methanol.[25]
Migratory Preferences
The ratio of the two possible carbonyl products (I and II) obtained is determined by the relative migratory abilities of the carbonyl substituents (R1 and R2). In general, the R-group most capable of stabilizing the partial positive charge formed during the rearrangement migrates preferentially. A prominent exception to this general rule is hydride shifting. The migratory preferences of the carbonyl R-groups can be heavily influenced by solvent and diazoalkane choice. For example, methanol has been shown to promote aryl migration.[11][12] As shown below, if the reaction of piperanol (IV) with diazomethane is carried out in the absence of methanol, the ketone obtained though a hydride shift is the major product (V). If methanol is the solvent, an aryl shift occurs to form the aldehyde (VI), which cannot be isolated as it continues to react to form the ketone (VII) and the epoxide (VIII) products.[11][12]

The diazoalkane employed can also determine relative yields of products by influencing migratory preferences, as conveyed by the reactions of o-nitropiperonal with diazomethane and diazoethane. In the reaction between o-nitropiperonal (IX) and diazomethane, an aryl shift leads to production of the epoxide (X) in 9 to 1 excess of the ketone product (XI). When diazoethane is substituted for diazomethane, a hydride shift produces the ketone (XII), the only isolable product.[26]

Examples in the Literature
The Büchner–Curtius–Schlotterbeck reaction can be used to facilitate one carbon ring expansions when the substrate ketone is cyclic. For instance, the reaction of cyclopentanone with diazomethane forms cyclohexanone (shown below). The Büchner ring expansion reactions utilizing diazoalkanes have proven to be synthetically useful as they can not only be used to form 5- and 6-membered rings, but also more unstable 7- and 8-membered rings.[27]

An acyl-diazomethane can react with an aldehyde to form a β-diketone in the presence of a transition metal catalyst (SnCl2 in the example shown below). β-Diketones are common biological products, and as such, their synthesis is relevant to biochemical research. Furthermore, the acidic β-hydrogens of β-diketones are useful for broader synthetic purposes, as they can be removed by common bases.[27]
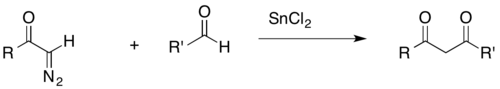
Acyl-diazomethane can also add to esters to form β-keto esters, which are important for fatty acid synthesis. As mentioned above, the acidic β-hydrogens also have productive functionality.[27]
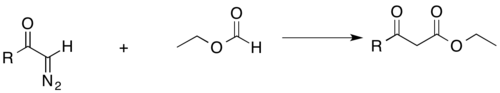
The Büchner–Curtius–Schlotterbeck reaction can also be used to insert a methylene bridge between a carbonyl carbon and a halogen of an acyl halide. This reaction allows conservation of the carbonyl and halide functionalities.[28]

It is possible to isolate nitrogen-containing compounds using the Büchner–Curtius–Schlotterbeck reaction. For example, an acyl-diazomethane can react with an aldehyde in the presence of a DBU catalyst to form isolable α-diazo-β-hydroxy esters (shown below).[27]
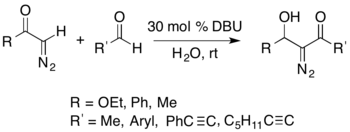
References
- ↑ "Büchner–Curtius–Schlotterbeck Reaction". Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons, Inc. Retrieved 2013-04-28.
- ↑ Bruchner, E; Theodore Curtius (1885). "Synthese von Ketonsäureäthern aus Aldehyden und Diazoessigäther". Berichte der Deutschen Chemischen Gesellschaft 18 (2): 2371–2377. doi:10.1002/cber.188501802118.
- 1 2 3 Schlotterbeck, Fritz (1907). "Transformation of Aldehydes into Ketones by Means of Diazomethane". Berichte der Deutschen Chemischen Gesellschaft 40: 479, 1826, 3000. doi:10.1002/cber.19070400285.
- ↑ von Pechmann, Hans (1895). "Ueber Diazomethan". Berichte der deutschen chemischen Gesellschaft 28 (1): 855–861. doi:10.1002/cber.189502801189.
- ↑ Meyer, Viktor (1905). Monatshefte für Chemie 26: 1295. Missing or empty
|title=(help) - ↑ Bandgar, B.; S. Pandit; V. Sadavarte (2001). "Montmorillonite K-10 catalyzed synthesis of β-keto esters: condensation of ethyl diazoacetate with aldehydes under mild conditions". Green Chemistry 3 (5): 247–249. doi:10.1039/B104116A.
- 1 2 3 4 Pohls, P. (1934). Inaugural Dissertation. Marburg, Germany: University of Marburg.
- 1 2 Pauli, O. (1935). Inaugural Dissertation. Marburg, Germany: University of Marburg.
- 1 2 C. David Gutsche (1954). "Volume 8, Chapter 8: The Reaction of Diazomethane and its Derivatives with Aldehydes and Ketones". In Roger Adams. Organic Reactions. New York: John Wiley & Sons. p. 396.
- ↑ Holmquist, Christopher; Eric Roskamp (24 April 1989). "A Selective Method for Diazoacetate Catalyzed the Direct Conversion of Aldehydes into β-Keto Esters with Ethyl by Tin(II) Chloride". Journal of Organic Chemistry 54: 3258–3260. doi:10.1021/jo00275a006.
- 1 2 3 4 5 6 7 Arndt, Fritz; J. Amende; W. Ender (1 March 1932). "Synthesen mit Diazomethan VII Weiteres über die Umsetzung von Aldehyden und Ketonen". Monatshefte Chemistry 59 (1-2): 203–220. doi:10.1007/BF01638230.
- 1 2 3 Mosettig, Erich (13 June 1928). "Über die Einwirkung von Diazo-methan auf Piperonal". Berichte der deutschen chemischen Gesellschaft (A and B Series) 61 (6): 1391–1395. doi:10.1002/cber.19280610634.
- 1 2 3 Eistert (1941). Angew. Chem. 54 (99): 124. Missing or empty
|title=(help) - ↑ Meerwin, Ger. pat. 579,309 [C. A., 27, 4546 (1933)]
- ↑ Kohler, E.; M. Tishler; H. Potter; H. Thompson (May 1939). "The Preparation of Cyclic Ketones by Ring Enlargement". Journal of the American Chemical Society 61 (5): 1057–1061. doi:10.1021/ja01874a021.
- ↑ Gutsche, C. (October 1949). "Ring Enlargements. I. The Ring Enlargement of 2-Chlorocyclohexanone and 2-Phenylcyclohexanone". Journal of the American Chemical Society 71 (10): 3513–3517. doi:10.1021/ja01178a075.
- ↑ Mousseron; Manon (1949). Bulletin de la Societe Chimique: 392. Missing or empty
|title=(help) - ↑ Mosettig, Erich; Alfred Burger (June 1931). "New Alkamines in the Tetrahydronaphthalene Series". Journal of the American Chemical Society 53 (6): 2295–2300. doi:10.1021/ja01357a038.
- ↑ Giraitis, Albert; Jesse Bullock (May 1937). "Reactions of Cyclohexanone with Diazoethane". Journal of the American Chemical Society 59 (5): 951. doi:10.1021/ja0128a511.
- ↑ Adamson, Donald W.; J. Kenner (1937). "Improved preparations of aliphatic diazo-compounds, and certain of their properties". Journal of the Chemical Society: 1551–1556. doi:10.1039/JR9370001551.
- ↑ Adamson, Donald W.; J. Kenner (1939). "Reactions of aliphatic diazo-compounds with carbonyl derivatives". Journal of the Chemical Society: 181–189. doi:10.1039/JR9390000181.
- ↑ Wilds, A.L.; Arthur L. Meader Jr. (September 1948). "The use of higher diazohydrocarbons in the Arndt–Eistert synthesis". Journal of Organic Chemistry 13 (5): 763–779. doi:10.1021/jo01163a024. PMID 18884425.
- ↑ Ramonczai, J.; L. Vargha (June 1950). "Studies of furan compounds. III. A new synthesis of furyl ketones". Journal of the American Chemical Society 72 (6): 2737. doi:10.1021/ja01162a109.
- ↑ Arndt, F.; Eistert, B.; Ender, W. (1929). "Synthesen mit diazo-methan, VI.: Uber die Reaktion von Ketonen und Aldehyden mit diazo-methan". Berichte der deutschen chemischen Gesellschaft 62 (1): 44–56. doi:10.1002/cber.19290620106.
- ↑ C. David Gutsche (1954). "Volume 8, Chapter 8: The Reaction of Diazomethane and its Derivatives with Aldehydes and Ketones". In Roger Adams. Organic Reactions. New York: John Wiley & Sons. pp. 369–370.
- ↑ Mosettig, Erich; Karl Czadek (1931). "Reactions of diazomethane with piperonal. III". Monatsheft fuer Chemie 57: 291–304.
- 1 2 3 4 Zhang, Yan; Jianbo Wang (22 July 2009). "Recent development of reactions with a -diazocarbonyl compounds as nucleophiles". The Royal Society of Chemistry: 5350–5361. doi:10.1039/b908378b.
- ↑ Fried, Josef; Robert C. Elderfield (July 1941). "STUDIES ON LACTONES RELATED TO THE CARDIAC AGLYCONES. VI. THE ACTION OF DIAZOMETHANE ON CERTAIN DERIVATIVES OF α-PYRONE". Journal of Organic Chemistry 6 (4): 577–583. doi:10.1021/jo01204a011.