Argininemia
| Argininemia | |
|---|---|
 | |
| Classification and external resources | |
| OMIM | 207800 |
| DiseasesDB | 29677 |
| eMedicine | ped/132 |
| MeSH | D020162 |
| GeneReviews | |
Argininemia, also called arginase deficiency,[1] is an autosomal recessive[2] urea cycle disorder where a deficiency of the enzyme arginase causes a buildup of arginine and ammonia in the blood.
Ammonia, which is formed when proteins are broken down in the body, is toxic if levels become too high. The nervous system is especially sensitive to the effects of excess ammonia.
Diagnosis
Argininemia usually becomes evident by about the age of 3. It most often appears as stiffness, especially in the legs, caused by abnormal tensing of the muscles (spasticity). Other symptoms may include slower than normal growth, developmental delay and eventual loss of developmental milestones, intellectual disability, seizures, tremor, and difficulty with balance and coordination (ataxia). Occasionally, high protein meals or stress caused by illness or periods without food (fasting) may cause ammonia to accumulate more quickly in the blood. This rapid increase in ammonia may lead to episodes of irritability, refusal to eat, and vomiting.
In some affected individuals, signs and symptoms of argininemia may be less severe, and may not appear until later in life.
Genetics
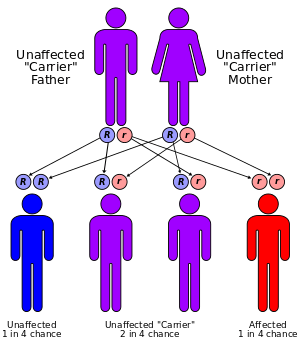
Mutations in the ARG1 gene cause argininemia.
Argininemia belongs to a class of genetic diseases called urea cycle disorders. The urea cycle is a sequence of reactions that occurs in liver cells. This cycle processes excess nitrogen, generated when protein is used by the body, making a compound called urea that is excreted by the kidneys.
The ARG1 gene provides instructions for making an enzyme called arginase. This enzyme controls the final step of the urea cycle, which produces urea by removing nitrogen from arginine. In people with arginase deficiency, arginase is damaged or missing, and arginine is not broken down properly. As a result, urea cannot be produced normally, and excess nitrogen accumulates in the blood in the form of ammonia. The accumulation of ammonia and arginine are believed to cause the neurological problems and other signs and symptoms of arginase deficiency. The accumulation of guanidino metabolites also contribute to the neurotoxicity of the disease.[3]
This condition is an autosomal recessive disorder, which means the defective gene is located on an autosome, and two copies of the defective gene are required to inherit the disorder. Both parents of an individual with an autosomal recessive disorder are carriers of one copy of the gene, but usually do not have the disorder.
References
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 207800
- ↑ Uchino, Takako; Snyderman, Selma E.; Lambert, Marie; Qureshi, Ijaz A.; Shapira, Stuart K.; Sansaricq, Claude; Smit, Leonard M. E.; Jakobs, Cornelis; Matsuda, Ichiro (1995). "Molecular basis of phenotypic variation in patients with argininemia". Human Genetics 96 (3): 255–60. doi:10.1007/BF00210403. PMID 7649538.
- ↑ Deignan, Joshua L.; Marescau, Bart; Livesay, Justin C.; Iyer, Ramaswamy K.; De Deyn, Peter P.; Cederbaum, Stephen D.; Grody, Wayne W. (2008). "Increased plasma and tissue guanidino compounds in a mouse model of hyperargininemia". Molecular Genetics and Metabolism 93 (2): 172–8. doi:10.1016/j.ymgme.2007.09.016. PMID 17997338.
External links
- GeneReviews/NCBI/NIH/UW entry on Urea Cycle Disorders Overview
- GeneReviews/NCBI/NIH/UW entry on Arginase Deficiency or Hyperargininemia
- OMIM entries on Arginase Deficiency
- Argininemia at NLM Genetics Home Reference
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||