Antibody-dependent enhancement
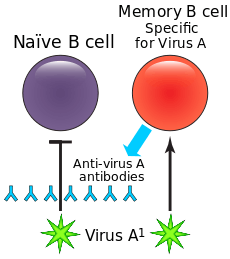
Antibody-dependent enhancement (ADE) occurs when non-neutralising antiviral proteins facilitate virus entry into host cells, leading to increased infectivity in the cells. Some cells do not have the usual receptors on their surfaces that viruses use to gain entry. The antiviral proteins (i.e., the antibodies) bind to antibody Fc receptors that some of these cells have in the plasma membrane. The viruses bind to the antigen binding site at the other end of the antibody. ADE is common in cells cultured in the laboratory, but rarely occurs in vivo except for dengue virus. This virus can use this mechanism to infect human macrophages, causing a normally mild viral infection to become life-threatening.[1]
In dengue virus infection
The most widely known example of ADE occurs in the setting of infection with the dengue virus (DENV). DENV is a single-stranded positive-polarity RNA virus of the Flaviviridae family. It causes a disease of varying severity in humans, from dengue fever (DF), which is usually self-limited, to dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS), either of which may be life-threatening.[2] It is estimated that as many as 390 million individuals are infected with DENV annually.[3]
The phenomenon of ADE may be observed when a person who has previously been infected with one serotype of DENV becomes infected many months or years later with a different serotype. In such cases, the clinical course of the disease is more severe, and these people have higher viremia compared with those in whom ADE has not occurred. This explains the observation that while primary (first) infections cause mostly minor disease (DF) in children, secondary infection (re-infection at a later date) is more likely to be associated with severe disease (DHF and/or DSS) in both children and adults.[4]
There are four antigenically different serotypes of DENV (DENV-1 - DENV-4).[5] Infection with DENV induces the production of neutralizing homotypic immunoglobulin G (IgG) antibodies which provide lifelong immunity against the infecting serotype. Infection with DENV also produces some degree of cross-protective immunity against the other three serotypes.[6] Neutralizing heterotypic (cross-reactive) IgG antibodies are responsible for this cross-protective immunity, which typically persists for a period of several months to a few years. These heterotypic antibody titers decrease over long time periods (4 to 20 years).[7] While heterotypic IgG antibody titers decrease, homotypic IgG antibody titers increase over long time periods. This could be due to the preferential survival of long-lived memory B cells producing homotypic antibodies.[7]
In addition to inducing neutralizing heterotypic antibodies, infection with DENV can also induce heterotypic antibodies which neutralize the virus only partially or not at all.[8] The production of such cross-reactive but non-neutralizing antibodies could be the reason for more severe secondary infections. It is thought that by binding to but not neutralizing the virus, these antibodies cause it to behave as a "trojan horse",[9][10][11] where it is delivered into the wrong compartment of dendritic cells that have ingested the virus for destruction.[12][13] Once inside the white blood cell, the virus replicates undetected, eventually generating very high virus titers which cause severe disease.[14]
A recent study conducted by Modhiran et al.[15] attempted to explain how non-neutralizing antibodies down regulate the immune response in the host cell through the TLR signaling pathway. TLR receptors are known to recognize extra and intra cellular viral particles and to be a major basis of the cytokines production. In vitro experiments showed that the inflammatory cytokines and Type 1 IFN production was reduced when the ADE-DENV complexe bound to the Fc receptor of THP-1 cells. This can be explained by both a decrease of TLR production and a modification of its signaling pathway. On one hand, an unknown protein induced by the stimulated Fc receptor reduces the TLRs transcription and translation, which reduce the capacity of the cell to detect viral proteins. On the other hand, many proteins (TRIF, TRAF6, TRAM, TIRAP, IKKα, TAB1, TAB2, NF-κB complex) involved in the TLR signaling pathway are down regulated, which lead to a decrease of the cytokine production. Two of them, TRIF and TRAF6, are respectively down regulated by 2 proteins SARM and TANK up regulated by the stimulated Fc receptors.
To illustrate the phenomenon of ADE, consider the following example: an epidemic of dengue fever occurred in Cuba, lasting from 1977 to 1979. The infecting serotype was DENV-1. This epidemic was followed by two more outbreaks of dengue fever—one in 1981 and one in 1997; DENV-2 was the infecting serotype in both of these later epidemics. 205 cases of DHF/DSS occurred during the 1997 outbreak, all in people older than 15 years. All but three of these cases were demonstrated to have been previously infected by the DENV-1 serotype during the epidemic of 1977–1979.[16] Furthermore, people who had been infected with DENV-1 during the 1977-79 outbreak and secondarily infected with DENV-2 in 1997 had a 3-4 fold increased probability of developing severe disease than those secondarily infected with DENV-2 in 1981.[7] This scenario can be explained by the presence of neutralizing heterotypic IgG antibodies in sufficient titers in 1981, the titers of which had decreased by 1997 to the point where they no longer provided significant cross-protective immunity.
In HIV-1 virus infection
ADE of infection has also been reported in HIV. Like DENV, non-neutralizing level of antibodies have been found to enhance the viral infection through interactions of complement system and receptors.[17] The increase in infection has been reported to be over 350 fold which is comparable to ADE in other viruses like DENV.[17] ADE in HIV can be complement mediated or Fc receptor mediated. Complements in presence of HIV-1 positive sera have been found to enhance the infection of MT-2 T-cell line. The Fc-receptor mediated enhancement was reported when HIV infection was enhanced by sera from HIV-1 positive guinea pig enhanced the infection of peripheral blood mononuclear cells without the presence of any complements.[18] Complement Component receptors CR2, CR3 and CR4 have been found to mediate this Complement –Mediated enhancement of infection.[17][19] The infection of HIV-1 leads to activation of complements fragments of these complements can assist viruses with infection by facilitating viral interactions with host cells that express complement receptors.[20] The deposition of complement on the virus brings the gp120 protein close to CD4 molecules on the surface of the cells, thus leading to facilitated viral entry.[20] Viruses pre-exposed to non-neutralizing complement system have also been found to enhance infections in interdigitating dendritic cells (iDCs). Opsonized viruses have not only shown enhanced entry but also favorable signalling cascades for HIV replication in iDCs.[21] HIV-1 has also showed enhancement of infection in HT-29 cells when the viruses were pre-opsonized with complements C3 and C9 in seminal fluid. This enhanced rate of infection was almost 2 times greater than infection of HT-29 cells with virus alone.[22] Subramanian et al., reported that almost 72% of serum samples out of 39 HIV positive individuals contained complements that were known to enhance the infection. They also suggested that presence of neutralizing antibody (NA) or antibody-dependent cellular cytotoxicity-mediating antibodies (ADCC) in the serum contains infection enhancing antibodies(IEAs).[23] The balance between the NAs and IEAs changes as the disease progresses. During advanced stages of disease the proportion of IEAs are generally higher than NAs.[24] Increase in viral protein synthesis and RNA production have been reported to occur during the complement mediated enhancement of infection. Cells that are challenged with non neutralizing levels of complements have been found have accelerated release of reverse transcriptase and the viral progeny.[25] The interaction of anti-HIV antibodies with non-neutralizing complement exposed viruses also aid in binding of the virus and the erythrocytes which can lead to a more efficient delivery of viruses to the immune compromised organs.[19]
ADE in HIV has raised questions about the risk of infections to volunteers who have taken subneutalizing levels of vaccine just like any other viruses that exhibit ADE. Gilbert et al., in 2005 reported that there was no ADE of infection when they used rgp120 vaccine in phase 1 and 2 trials.[26] It has been emphasized that much research needs to be done in the field of immunity response to HIV-1, information from these studies can be used to produce a more effective vaccine.
Mechanism
There are several possibilities to explain the phenomenon:
- A viral surface protein laced with antibodies against a virus of one serotype binds to a similar virus with a different serotype. The binding is meant to neutralize the virus surface protein from attaching to the cell, but the antibody bound to virus also binds to the receptor of the cell, the Fc-region antibody receptor FcγR. This brings the virus into close proximity to the virus-specific receptor, and the cell internalizes the virus through the normal infection route.[27]
- A virus surface protein may be attached to antibodies of a different serotype, activating the classical pathway of the complement system. The complement cascade system instead binds C1Q complex attached to the virus surface protein via the antibodies, which in turn bind C1q receptor found on cells, bringing the virus and the cell close enough for a specific virus receptor to bind the virus, beginning infection. This mechanism has not been shown specifically for DENV infection, but is supposed to occur with Ebola virus infection in vitro.[28]
- When an antibody to a virus is present for a different serotype, it is unable to neutralize the virus, which is then ingested into the cell as a sub-neutralized virus particle. These viruses are phagocytosed as antigen-antibody complexes, and degraded by macrophages. Upon ingestion the antibodies no longer even sub-neutralize the body due to the denaturing condition at the step for acidification of phagosome before fusion with lysosome. The virus becomes active and begins its proliferation within the cell.
See also
- Original antigenic sin
- Influenza A virus subtype H7N9 where this is suspected.
References
- ↑ Dimmock, N. J.; Easton, A. J. (Andrew J.); Leppard, Keith. (2007). Introduction to modern virology. Malden, MA: Blackwell Pub. p. 65. ISBN 978-1-4051-3645-7.
- ↑ Boonnak, K; Slike, BM; Burgess, TH; Mason, RM; Wu, SJ; Sun, P; Porter, K; Rudiman, IF; et al. (2008). "Role of Dendritic Cells in Antibody-Dependent Enhancement of Dengue Virus Infection". Journal of Virology 82 (8): 3939–51. doi:10.1128/JVI.02484-07. PMC 2292981. PMID 18272578.
- ↑ Stinchcomb, Dan; Ambuel, Yuping; Young, Ginger; Brewoo, Joeseph; Paykal, Joanna; Weisgrau, Kim; Rakasz, Eva; Haller, Aurelia; Royals, Michael; Capuano, Saverio; Partidos, Charalambos; Osorio, Jorge (15 Sep 2014). "A Rapid Immunization Strategy with a Live-Attenuated Tetravalent Dengue Vaccine Elicits Protective Neutralizing Antibody Responses in Non-Human Primates". Frontiers in Immunology 5 (2014): 436. doi:10.1038/sj.emboj.7600199. PMC 404319. PMID 15057269.
- ↑ Guzman MG, Vazquez S (December 2010). "The complexity of antibody-dependent enhancement of dengue virus infection". Viruses 2 (12): 2649–62. doi:10.3390/v2122649. PMID 21994635.
- ↑ King, CA; Anderson, R; Marshall, JS (2002). "Dengue Virus Selectively Induces Human Mast Cell Chemokine Production". Journal of Virology 76 (16): 8408–19. doi:10.1128/JVI.76.16.8408-8419.2002. PMC 155122. PMID 12134044.
- ↑ Alvarez, G; Piñeros, JG; Tobón, A; Ríos, A; Maestre, A; Blair, S; Carmona-Fonseca, J (2006). "Efficacy of three chloroquine-primaquine regimens for treatment of Plasmodium vivax malaria in Colombia". The American journal of tropical medicine and hygiene 75 (4): 605–9. PMID 17038680.
- 1 2 3 Guzman, MG; Alvarez, M; Rodriguez-Roche, R; Bernardo, L; Montes, T; Vazquez, S; Morier, L; Alvarez, A; et al. (2007). "Neutralizing Antibodies after Infection with Dengue 1 Virus". Emerging Infectious Diseases 13 (2): 282–6. doi:10.3201/eid1302.060539. PMC 2725871. PMID 17479892.
- ↑ Goncalvez, AP; Engle, RE; St. Claire, M; Purcell, RH; Lai, CJ (2007). "Monoclonal antibody-mediated enhancement of dengue virus infection in vitro and in vivo and strategies for prevention". Proceedings of the National Academy of Sciences 104 (22): 9422–7. Bibcode:2007PNAS..104.9422G. doi:10.1073/pnas.0703498104. PMC 1868655. PMID 17517625.
- ↑ Peluso R, Haase A, Stowring L, Edward M, Ventura P (1985). "A Trojan mechanism for the spread of visna virus in monocytes". Virology 147 (1): 231–6. doi:10.1016/0042-6822(85)90246-6. PMID 2998068.
- ↑ Chen Y, Wang S (2002). "Activation of Terminally Differentiated Human Monocytes/Macrophages by Dengue Virus: Productive Infection, Hierarchical Production of Innate Cytokines and Chemokines, and the Synergistic Effect of Lipopolysaccharide". J Virol 76 (19): 9877–87. doi:10.1128/JVI.76.19.9877-9887.2002. PMC 136495. PMID 12208965. Retrieved 2011-05-01.
- ↑ Witayathawornwong P (2005). "Case report: fatal dengue encephalitis" (PDF). Southeast Asian J Trop Med Public Health 36 (1): 200–2. ISSN 0125-1562. PMID 15906668. Retrieved 2011-05-01.
- ↑ Rodenhuis-Zybert IA, Wilschut J, Smit JM (August 2010). "Dengue virus life cycle: viral and host factors modulating infectivity". Cell. Mol. Life Sci. 67 (16): 2773–86. doi:10.1007/s00018-010-0357-z. PMID 20372965.
- ↑ Guzman MG, Halstead SB, Artsob H; et al. (December 2010). "Dengue: a continuing global threat". Nat. Rev. Microbiol. 8 (12 Suppl): S7–S16. doi:10.1038/nrmicro2460. PMID 21079655.
- ↑ Dejnirattisai W, Jumnainsong A, Onsirisakul N; et al. (May 2010). "Cross-reacting antibodies enhance dengue virus infection in humans". Science 328 (5979): 745–8. Bibcode:2010Sci...328..745D. doi:10.1126/science.1185181. PMID 20448183.
- ↑ Naphak Modhiran, Siripen Kalayanarooj, Sukathida Ubol (2010). "Subversion of Innate Defenses by the Interplay between DENV and Pre-Existing Enhancing Antibodies: TLRs Signaling Collapse". PLoS Neglected Tropical Diseases (PLOS ONE) 4: e924. doi:10.1371/journal.pntd.0000924.
- ↑ Guzman, MG (2000). "Dr. Guzman et al. Respond to Dr. Vaughn". American Journal of Epidemiology 152 (9): 804. doi:10.1093/aje/152.9.804.
- 1 2 3 Willey, S; Aasa-Chapman, MI; O'Farrell, S; Pellegrino, P; Williams, I; Weiss, RA; Neil, SJD (2011). "Extensive complement-dependent enhancement of HIV-1 by autologous non-neutralising antibodies at early stages of infection". Retrovirology 8: 16. doi:10.1186/1742-4690-8-16. PMC 3065417. PMID 21401915.
- ↑ Levy, JA (2007). HIV and the pathogenesis of AIDS. Wiley-Blackwell. p. 247. ISBN 1-55581-393-3.
- 1 2 Yu, Q; Yu, R; Qin, X (2010). "The good and evil of complement activation in HIV-1 infection". Cellular and Molecular Immunology 7 (5): 334–40. doi:10.1038/cmi.2010.8. PMID 20228834.
- 1 2 Gras, GS; Dormont, D (1991). "Antibody-dependent and antibody-independent complement-mediated enhancement of human immunodeficiency virus type 1 infection in a human, Epstein-Barr virus-transformed B-lymphocytic cell line". Journal of Virology 65 (1): 541–5. PMC 240554. PMID 1845908.
- ↑ Bouhlal, H; Chomont, N; Réquena, M; Nasreddine, N; Saidi, H; Legoff, J; Kazatchkine, MD; Bélec, L; Hocini, H (2007). "Opsonization of HIV with Complement Enhances Infection of Dendritic Cells and Viral Transfer to CD4 T Cells in a CR3 and DC-SIGN-Dependent Manner". Journal of Immunology 178 (2): 1086–95. doi:10.4049/jimmunol.178.2.1086. PMID 17202372.
- ↑ Bouhlal, H; Chomont, N; Haeffner-Cavaillon, N; Kazatchkine, MD; Belec, L; Hocini, H (2002). "Opsonization of HIV-1 by Semen Complement Enhances Infection of Human Epithelial Cells". Journal of Immunology 169 (6): 3301–6. doi:10.4049/jimmunol.169.6.3301. PMID 12218150.
- ↑ Subbramanian, RA; Xu, J; Toma, E; Morisset, R; Cohen, EA; Menezes, J; Ahmad, A (2002). "Comparison of Human Immunodeficiency Virus (HIV)-Specific Infection-Enhancing and -Inhibiting Antibodies in AIDS Patients". Journal of Clinical Microbiology 40 (6): 2141–6. doi:10.1128/JCM.40.6.2141-2146.2002. PMC 130693. PMID 12037078.
- ↑ Beck, Z; Prohászka, Z; Füst, G (2008). "Traitors of the immune system—Enhancing antibodies in HIV infection: Their possible implication in HIV vaccine development". Vaccine 26 (24): 3078–85. doi:10.1016/j.vaccine.2007.12.028. PMID 18241961.
- ↑ Robinson Jr, WE; Montefiori, DC; Mitchell, WM (1990). "Complement-mediated antibody-dependent enhancement of HIV-1 infection requires CD4 and complement receptors". Virology 175 (2): 600–4. doi:10.1016/0042-6822(90)90449-2. PMID 2327077.
- ↑ Gilbert, PB; Peterson, ML; Follmann, D; Hudgens, MG; Francis, DP; Gurwith, M; Heyward, WL; Jobes, DV; et al. (2005). "Correlation between Immunologic Responses to a Recombinant Glycoprotein 120 Vaccine and Incidence of HIV‐1 Infection in a Phase 3 HIV‐1 Preventive Vaccine Trial". The Journal of Infectious Diseases 191 (5): 666–77. doi:10.1086/428405. PMID 15688279.
- ↑ Takada, A; Kawaoka, Y (2003). "Antibody-dependent enhancement of viral infection: molecular mechanisms and in vivo implications". Reviews in Medical Virology 13 (6): 387–98. doi:10.1002/rmv.405. PMID 14625886.
- ↑ Takada, A; Feldmann, H; Ksiazek, TG; Kawaoka, Y (2003). "Antibody-Dependent Enhancement of Ebola Virus Infection". Journal of Virology 77 (13): 7539–44. doi:10.1128/JVI.77.13.7539-7544.2003. PMC 164833. PMID 12805454.