Carbonyl reduction

In organic chemistry, carbonyl reduction is the organic reduction of any carbonyl group by a reducing agent.
Typical carbonyl compounds are ketones, aldehydes, carboxylic acids, esters, and acid halides. Carboxylic acids, esters, and acid halides can be reduced to either aldehydes or a step further to primary alcohols, depending on the strength of the reducing agent; aldehydes and ketones can be reduced respectively to primary and secondary alcohols. In deoxygenation, the alcohol can be further reduced and removed altogether.
Metal hydrides based on boron and aluminum are common reducing agents; catalytic hydrogenation is also an important method of reducing carbonyls. Before the discovery of soluble hydride reagents, esters were reduced by the Bouveault–Blanc reduction, employing a mixture of sodium metal in the presence of alcohols.[1][2]
Carboxylic acid derivatives, aldehydes, and ketones to alcohols
Hydride reduction
Mechanism
The reaction mechanism for metal hydride reduction is based on nucleophilic addition of hydride to the carbonyl carbon. In some cases, the alkali metal cation, especially Li+, activates the carbonyl group by coordinating to the carbonyl oxygen, thereby enhancing the electrophilicity of the carbonyl.
For reductions of carboxylic acid derivatives, after reduction by an aluminium hydride ion, an elimination leads to the aldehyde product (which can be reduced a second time to an alcohol):

For reductions of aldehydes and ketones, an aluminium hydride ion reduces the compound to form an alkoxide salt. After the complete reduction, the alkoxide is protonated to give the alcohol product:

Trends in carbonyl reactivity
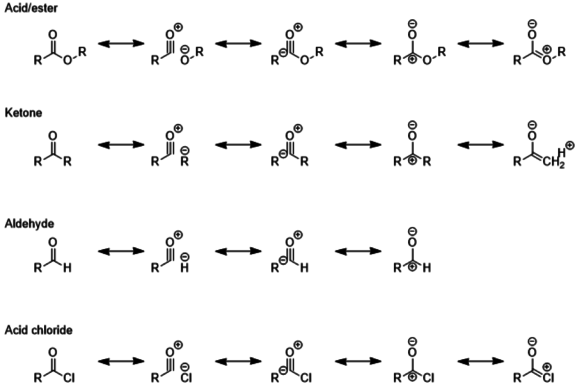
Ketones are less reactive than aldehydes, because of greater steric effects, and because the extra alkyl group can donate electron density to the partial positive charge of the polar C=O bond.[3] Therefore, aldehydes reduce more easily than ketones and require milder reagents and milder conditions. Carboxylic acids and esters are further stabilized by the presence of a second oxygen atom which can donate a lone pair into the already polar C=O bond. Acyl halides are the least stable of the carbonyls since halides are poor electron donors, as well as poor leaving groups.[4]
The result of these trends in carbonyl reactivity is that acid halides, ketones, and aldehydes are usually the most readily reduced compounds, while acids and esters require stronger reducing agents.
Trends in metal hydride reactivity
Four major factors contribute to the strength of metal hydride reducing agents. First, the counter ion’s ability to activate carbonyls depends on how well it can coordinate to the carbonyl oxygen. Lithium is smaller and more electrophilic than sodium, so it coordinates much more strongly and activates the carbonyl more.[5] Metals that can have multiple charges (such as Mg, Al, and Zn) form cations with high charge density, and are therefore also stronger activators than Na+.[6]
Second, the central metal can influence a reducing agent’s strength. Aluminum is larger than boron, so it bonds more weakly to hydrides, which are more free to attack; aluminum hydrides are therefore better reducers than borohydrides.[7] A third factor, sterics, is what makes certain substituted hydrides (hydrides in which one or more hydrides are replaced by substituents) much weaker reducers than other metal hydrides: sodium triacetoxyborohydride (NaBH(OAc)3), for instance, can be used to selectively reduce aldehydes, and leave the less reactive ketones unreacted.[8]
Finally, substituents can have other effects on a reducing agent’s reactivity: acetoxy groups hinder the reducing power of NaBH(OAc)3 not only through steric bulk but also because they are electron-withdrawing. Cyano groups also hinder reducing agents, while electron-donating groups such as alkyl groups can improve them, such as in superhydride (lithium triethylborohydride), which is a strong enough nucleophile to prevent undesired rearrangements during reduction.
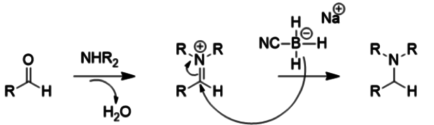
Because of these substituent effects, NaBH3CN is a very poor reducer at moderate pH (>4), so it prefers reductive amination to carbonyl reduction, as shown below:
The relatively weak reducer sodium borohydride is typically used for reducing ketones and aldehydes because unlike lithium aluminum hydride, it tolerates many functional groups (nitro group, nitrile, ester) and can be used with water or ethanol as solvents.[9] Lithium aluminum hydride and other strong reducers such as diisobutylaluminium hydride, L-selectride, diborane, diazene, and aluminum hydride can also reduce aldehydes and ketones, but are disfavored because they are hazardous and violently reactive.[10] However, these compounds are useful for reducing carboxylic acids and esters to alcohols, since sodium borohydride is not powerful enough to do so.
The following table illustrates which carbonyl functional groups can be reduced by which reducing agents (some of these reagents vary in efficacy depending on reaction conditions):

Carboxylic acid derivatives to aldehydes
Using metal hydrides
Forming aldehydes from carboxylic acid derivatives is often a challenge, because weaker reducing agents (NaBH4) are incapable of reducing esters and carboxylic acids, which are relatively stable, and stronger reducing agents (LiAlH4) immediately reduce the formed aldehyde to an alcohol.[11]
Since acid chlorides are less stable than aldehydes and ketones, they are often used in conjunction with sterically hindered hydride donors when synthesizing aldehydes, because the relatively weak reducer will react preferentially with the acid chloride starting material, leaving the aldehyde product unreacted. The reducing agent DIBAL-H (Diisobutylaluminium hydride) is often used for this purpose: though it normally reduces all carbonyls, it can stop reducing at the aldehyde if only one equivalent is used at low temperatures.[12] LiAl(OtBu)3 (formed from LiAlH4 and tBuOH in situ) can also stop reducing at the aldehyde, through a similar mechanism to DIBAL-H.[13]
Alternative methods
The traditional method of forming aldehydes without reducing to alcohols - by using hindered hydrides and reactive carbonyls - is limited by its narrow substrate scope and great dependence on reaction conditions. One workaround to avoid this method is to reduce the carboxylic acid derivative all the way down to an alcohol, then oxidize the alcohol back to an aldehyde. Other alternatives include forming a thioester or a Weinreb amide, then reducing the new species to an aldehyde through the Fukuyama reduction or Weinreb reaction respectively, or using catalytic hydrogenation as in the Rosenmund reaction.
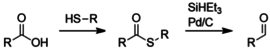
In the Fukuyama reduction, a carboxylic acid is first converted to a thioester through addition of a thiol (with a mechanism similar to esterification).[14] The thioester is then reduced to an aldehyde by a silyl hydride with a palladium catalyst.
In the Weinreb reaction, an acyl chloride is first converted to the Weinreb amide, then treated with an organometallic reagent to form a ketone, or lithium aluminum hydride to form an aldehyde:[15]

The Weinreb amide is reduced via a stable chelate, rather than the electrophilic carbonyl that is formed through metal hydride reductions; the chelate is therefore only reduced once, as illustrated below:

The Rosenmund reaction reduces acyl chlorides to aldehydes using hydrogen gas with a catalyst of palladium on barium sulfate, whose small surface area prevents over-reduction.[16] For more reactive substrates, the catalyst must be further hindered with a poison, often one containing sulfur.
Aldehydes and ketones to alkanes
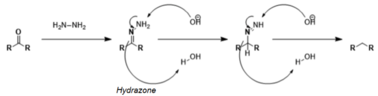
Aldehydes and ketones can be reduced not only to alcohols but also to alkanes. Some reactions for this transformation include the Clemmensen reduction (in strongly acidic conditions) and the Wolff-Kishner reduction (in strongly basic conditions), as well as the various modifications of the Wolff-Kishner reaction. The Caglioti modification, for instance, uses tosylhydrazone with a hydride donor in milder conditions with no base;[17] the Myers modification substitutes hydrazine with bis(tert-butyldimethylsilyl)-hydrazine, uses milder conditions at room temperature, and is rapid and efficient.[18]
α,β-unsaturated carbonyls
In α,β-reduction (also called conjugate reduction), the substrate is an α,β-unsaturated carbonyl, an enone or enal.
When these substrates are reduced, 1,2-reduction - which produces an allyl alcohol - is in competition with the 1,4-reduction - which forms the saturated ketone or aldehyde. The following NaBH4 reduction of an enone shows two possible products: the first from 1,4-reduction and the second from 1,2-reduction.[9]

The more sterically hindered the enone substrate, the more likely 1,2 reduction becomes.[9] Additionally, to selectively form the alcohol and avoid the 1,4 product, the Luche reaction uses the smaller molecule Ce(BH4)3 (derived from NaBH4 and CeCl3 combined in situ) as the hydride source.[19]
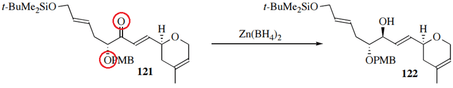
The hydride source Zn(BH4)2 also shows 1,2 selectivity, as well as greater diastereoselectivity; it does so by coordinating not only to the carbonyl oxygen but also to adjacent atoms:[20]
Stereoselectivity
Diastereoselective reduction
In the reduction of cyclohexanones, the hydride source can attack axially to produce an equatorial alcohol, or equatorially to produce an axial alcohol. In axial attack (shown in red), the hydride encounters 1,3-diaxial strain. In equatorial attack (shown in blue), the hydride avoids the 1,3-diaxial interaction, but the substrate undergoes unfavorable torsional strain when the newly formed alcohol and added hydrogen atom eclipse each other in the reaction intermediate (as shown in the Newman projection for the axial alcohol).
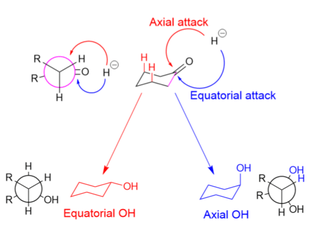
Large reducing agents, such as LiBH(Me2CHCHMe)3, are hindered by the 1,3-axial interactions and therefore attack equatorially.[9] Small reducing agents, such as NaBH4, preferentially attack axially in order to avoid the eclipsing interactions, because the 1,3-diaxial interaction for small molecules is minimal; stereoelectronic reasons have also been cited for small reducing agents' axial preference.[21] Making the substrate bulkier (and increasing 1,3-axial interactions), however, decreases the prevalence of axial attacks, even for small hydride donors.[22]
Enantioselective reduction
When asymmetrical ketones are reduced, the resulting secondary alcohol has a chiral center whose ccan be controlled using chiral catalysts.
Well-known carbonyl reductions in asymmetric synthesis are the Noyori asymmetric hydrogenation (beta-ketoester reduction/Ru/BINAP) and the CBS reduction (BH3, proline derived chiral catalyst).
References
- ↑ Organic Syntheses, Coll. Vol. 6, p.781 (1988); Vol. 53, p.70 (1973). Link
- ↑ Organic Syntheses, Coll. Vol. 4, p.834 (1963); Vol. 33, p.82 (1953). link
- ↑ Roche, Alex. "Ketones and Aldehydes" (PDF). Rutgers University. Retrieved December 1, 2015.
- ↑ Clayden, Jonathan (2012). Organic Chemistry. OUP Oxford. p. 200. ISBN 0199270295.
- ↑ König, Burkhard (2009). "Reduction Reactions" (PDF). Modern Methods in Organic Synthesis. Institut für Organische Chemie, Uni Regensburg. Retrieved December 1, 2015.
- ↑ Cox, Liam (2007). "Nucleophilic Addition Reactions of Aldehydes and Ketones" (PDF). University of Birmingham. Retrieved December 1, 2015.
- ↑ Sweeting, Linda M. (2001). "Reducing Agents". Towson University. Retrieved December 1, 2015.
- ↑ Gribble, Gordon W.; Ferguson, Duncan C. "Reactions of sodium borohydride in acidic media. Selective reduction of aldehydes with sodium triacetoxyborohydride". pubs.rsc.org. doi:10.1039/C39750000535. Retrieved 2015-12-02.
- 1 2 3 4 Banfi, Luca; Narisano, Enrica; Riva, Renata (2001-01-01). Sodium Borohydride. John Wiley & Sons, Ltd. doi:10.1002/047084289x.rs052. ISBN 9780470842898.
- ↑ Chaikin, Saul W.; Brown, Weldon G. (1949-01-01). "Reduction of Aldehydes, Ketones and Acid Chlorides by Sodium Borohydride". Journal of the American Chemical Society 71 (1): 122–125. doi:10.1021/ja01169a033. ISSN 0002-7863.
- ↑ Gaylord, Norman G. (1957-08-01). "Reduction with complex metal hydrides". Journal of Chemical Education 34 (8). doi:10.1021/ed034p367.
- ↑ Zakharkin, L.I.; Khorlina, I.M. "Reduction of esters of carboxylic acids into aldehydes with diisobutylaluminium hydride". Tetrahedron Letters 3 (14): 619–620. doi:10.1016/s0040-4039(00)70918-x.
- ↑ Cortes, Sergio (2010). "Using Hydrogen as a Nucleophile in Hydride Reductions" (PDF). Dr. Sergio Cortes' Organic Chemistry Page. University of Texas at Dallas. Retrieved December 1, 2015.
- ↑ Fukuyama, Tohru; Lin, Shao Cheng; Li, Leping (1990-09-01). "Facile reduction of ethyl thiol esters to aldehydes: application to a total synthesis of (+)-neothramycin A methyl ether". Journal of the American Chemical Society 112 (19): 7050–7051. doi:10.1021/ja00175a043. ISSN 0002-7863.
- ↑ Nahm, Steven; Weinreb, Steven M. "N-methoxy-n-methylamides as effective acylating agents". Tetrahedron Letters 22 (39): 3815–3818. doi:10.1016/s0040-4039(01)91316-4.
- ↑ Mosettig, Erich; Mozingo, Ralph (2004-01-01). The Rosenmund Reduction of Acid Chlorides to Aldehydes. John Wiley & Sons, Inc. doi:10.1002/0471264180.or004.07. ISBN 9780471264187.
- ↑ Caglioti, L.; Magi, M. (1963-01-01). "The reaction of tosylhydrazones with lithium aluminium hydride". Tetrahedron 19 (7): 1127–1131. doi:10.1016/S0040-4020(01)98571-0.
- ↑ Furrow, Michael E.; Myers, Andrew G. (2004-05-01). "Practical Procedures for the Preparation of N-tert-Butyldimethylsilylhydrazones and Their Use in Modified Wolff−Kishner Reductions and in the Synthesis of Vinyl Halides and gem-Dihalides". Journal of the American Chemical Society 126 (17): 5436–5445. doi:10.1021/ja049694s. ISSN 0002-7863.
- ↑ Strategic Applications of Named Reactions in Organic Synthesis (Paperback) by Laszlo Kurti, Barbara Czako ISBN 0-12-429785-4
- ↑ Greeves, Nick (2015). "Diastereoselective Ketone Reduction". ChemTube3D. University of Liverpool. Retrieved December 1, 2015.
- ↑ Wong, Stephen S.; Paddon-Row, Michael N. "Theoretical evidence in support of the Anh?Eisenstein electronic model in controlling ?-facial stereoselectivity in nucleophilic additions to carbonyl compounds". Journal of the Chemical Society, Chemical Communications (6). doi:10.1039/c39900000456.
- ↑ Krishnamurthy, S.; Brown, Herbert C. (1976-05-01). "Lithium trisiamylborohydride. A new sterically hindered reagent for the reduction of cyclic ketones with exceptional stereoselectivity". Journal of the American Chemical Society 98 (11): 3383–3384. doi:10.1021/ja00427a061. ISSN 0002-7863.