2,3-Wittig rearrangement
The [2,3]-Wittig rearrangement is the transformation of an allylic ether into a homoallylic alcohol via a concerted, pericyclic process. Because the reaction is concerted, it exhibits a high degree of stereocontrol, and can be employed early in a synthetic route to establish stereochemistry. The Wittig rearrangement requires strongly basic conditions, however, as a carbanion intermediate is essential. [1,2]-Wittig rearrangement is a competitive process.[1]
Introduction
[2,3]-Sigmatropic rearrangements occur for a variety of groups X and Y (see below). When X is a carbanion and Y an alkoxide, the rearrangement is called the [2,3]-Wittig rearrangement and the products are pent-1-en-5-ols. The [1,2]-Wittig rearrangement, which produces isomeric pent-5-en-1-ols, is a competitive process that takes place at high temperatures.[2] Because of the high atom economy and stereoselectivity of the [2,3]-rearrangement, it has gained considerable synthetic utility. The carbanion is generated by direct lithiation of moderately acidic substrates, tin transmetallation, or reductive lithiation of O,S-acetals. Stereoselective methods employing chiral starting materials have been used to effect either asymmetric induction or simple diastereoselection[3]
(1)
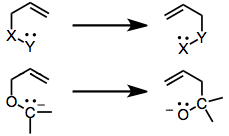
Mechanism and stereochemistry
Prevailing mechanism
After carbanion formation, the [2,3]-Wittig rearrangement is rapid and selective at low temperatures. However, if the reaction mixture is allowed to reach temperatures above −60 °C, [1,2]-rearrangement becomes competitive.[4]
(2)
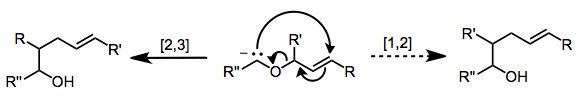
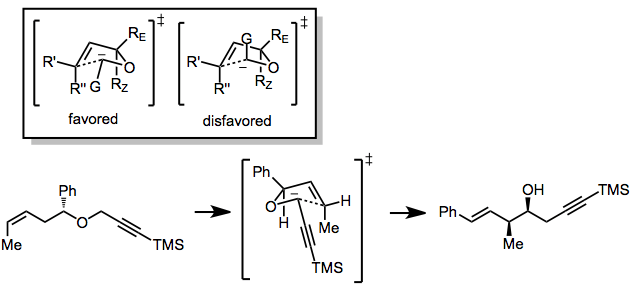
The postulated transition state possesses a five-membered, envelope-like structure.[5] The group attached to the carbanion (G) can occupy either a pseudoequatorial or pseudoaxial position, although the former is usually preferred. Large substituents on the other side of the ether oxygen prefer to occupy the exo position (RE) to avoid A1,3 strain. These restrictions lead to a preference for the syn product from (Z) isomers and anti products from (E) isomers; however, some exceptions to this rule are known.[6]
(3)
Stereoselective variants
Stereoselective variants of the [2,3]-Wittig rearrangement have employed three strategies: diastereoselection based on an existing, established stereocenter, placement of a chiral auxiliary on the starting material whose configuration is unaffected by the reaction, and the use of a chiral base. The relative diastereoselection strategy works well only for a limited number of G groups, but usually results in high yields because no chiral auxiliary group needs to be removed or modified. The stereocenter opposite the carbanion usually must be tertiary (rather than quaternary) in order to enforce the placement of the largest substituent in the RE position.[7]
(4)
The asymmetric induction approach relies on stereocenters already set in the starting material that are unaffected by the reaction (chiral auxiliaries). The most success has been achieved by placing these stereocenters either in the G group[8] or in a substituent attached to the terminal end of the double bond.[9] Diastereomeric ratios in excess of 90:10 are common for these reactions; however, removal of the chiral auxiliary is sometimes difficult.[10]
(5)
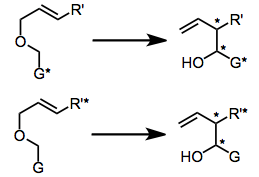
The use of chiral bases has afforded enantioenriched rearrangement products in a few cases,[11] although this method does not appear to be general. Enantioselectivity in these reactions is often low, suggesting that the association between the conjugate acid of the base and the rearranging carbanion is likely weak.
(6)
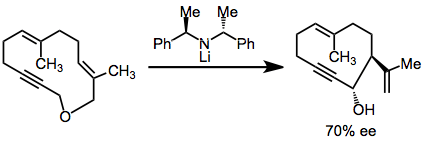
Scope and limitations
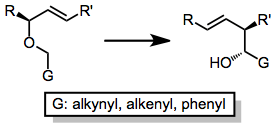
A variety of allylic ethers undergo the Wittig rearrangement—the fundamental requirement is the ability to generate the appropriate carbanion in the substrate. This demands either acidic hydrogens, a reducible functional group, or a carbon-metal bond. Historically, alkenyl, alkynyl, and phenyl groups have been used to acidify the α position. Free terminal alkynes are tolerated, although yields are higher when silyl-protected alkynes are used.[12]
(7)
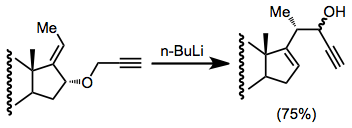
When an alkene is used as the anion-stabilizing group G, issues of selectivity arise concerning the site of the carbanion. Anion-stabilizing groups such as (trimethyl)silyl or methylthio provide essentially complete site selectivity.[13]
(8)
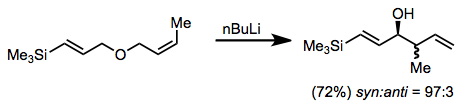
Carbonyl groups may also be used as the anion-stabilizing group; carbonyl groups are particularly useful for asymmetric rearrangements that employ chiral auxiliaries.[14]
(9)
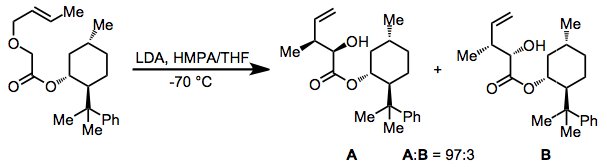
A highly enantioselective method employing chromium carbonyl complexes involves the use of the acidified phenyl ring as an anion-stabilizing group.[15]
(10)
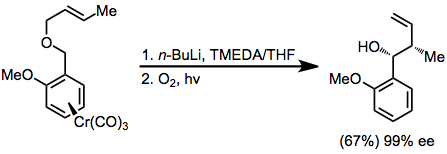
That the substrate must contain acidic hydrogens adjacent to the ether oxygen was a significant limitation of the original reaction. Thus, the development of transmetallation methods that allowed the selective generation of carbanions from carbon-tin bonds represented a profound methodological advance. The scope of the groups that could be attached to the anionic center expanded dramatically as a result.[6]
(11)

Synthetic applications
The products of the [2,3]-Wittig rearrangement of bis(allylic) ethers are 1,5-dien-3-ols. These substrates may undergo the oxy-Cope rearrangement upon deprotonation, affording δ,ε-unsaturated carbonyls. This tandem sigmatropic strategy has been employed in the synthesis of some natural products, including brevicomine and oxocrinal.[16]
(12)
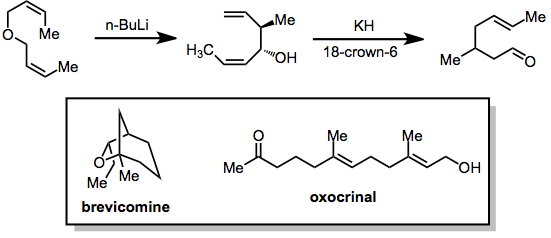
Experimental conditions and procedure
Typical conditions
Rearrangements must be carried out at temperatures below −60 °C to avoid competitive [1,2]-rearrangement. Typically, simple treatment of the substrate with n-butyllithium is sufficient to cause rearrangement. Reactions involving butyllithium should be carried out under nitrogen or argon with strict exclusion of water.
See also
References
- ↑ Nakai, T.; Mikami, K. (2004). "The [2,3]-Wittig Rearrangement". Organic Reactions. doi:10.1002/0471264180.or046.02. ISBN 0471264180.
- ↑ Baldwin, J. E.; Patrick, J. E. (1971). "Stereochemistry of [2,3]-sigmatropic reactions. Wittig rearrangement". J. Am. Chem. Soc. 93 (14): 3556. doi:10.1021/ja00743a060.
- ↑ Nakai, T.; Mikami, K.; Taya, S.; Fujita, Y. (1981). "[2,3]-Wittig rearrangement of unsymmetrical bis-allylic ethers. Facile method for regio- and stereoselective synthesis of 1,5-dien-3-ols". J. Am. Chem. Soc. 103 (21): 6492. doi:10.1021/ja00411a038.
- ↑ Schollkopf, U.; Fellenberger, K.; Rizk, M. (1970). "1.2-Wanderungen zum Atom mit freiem Elektronenpaar, VIII.ortho-Isomerisation bei anionisierten Äthern und Wanderungsmechanismus eines Propargyl-Restes bei der Wittig-Umlagerung". Justus Liebigs Ann. Chem. 734: 106. doi:10.1002/jlac.19707340111.
- ↑ Mikami, K.; Kimura, Y.; Kishi, N.; Nakai, T. (1983). "Acyclic diastereoselection of the [2,3]-Wittig sigmatropic rearrangement of a series of isomeric crotyl ethers. A conceptual model for the transition-state geometry". J. Org. Chem. 48 (2): 279. doi:10.1021/jo00150a033.
- 1 2 Still, W. C.; Mitra, A. (1978). "A highly stereoselective synthesis of Z-trisubstituted olefins via [2,3]-sigmatropic rearrangement. Preference for a pseudoaxially substituted transition state". J. Am. Chem. Soc. 100 (6): 1927. doi:10.1021/ja00474a049.
- ↑ Sayo, N.; Azuma, K.; Mikami, K.; Nakai, T. (1984). "Acyclic stereocontrol via asymmetric [2,3]-Wittig rearrangement with high enantio- and erythro-selectivity and its use in the chiral synthesis of insect pheromones". Tetrahedron Lett. 25 (5): 565. doi:10.1016/S0040-4039(00)99939-8.
- ↑ Mikami, K.; Fujimoto, K.; Kasuga, T.; Nakai, T. (1984). "Asymmetric [2,3]Wittig sigmatropic rearrangement involving a chiral azaenolate as the migrating terminus. A simple synthesis of (+)-verrucarinolactone". Tetrahedron Lett. 25 (52): 6011. doi:10.1016/S0040-4039(01)81746-9.
- ↑ Priepke, H.; Bruckner, R.; Harms, K. (1990). "Asymmetric Induction in the Wittig-Still Rearrangement of Ethers Containing an Allylic Stereocenter – Diastereocontrol by Allylic Nitrogen". Chem. Ber. 123 (3): 555. doi:10.1002/cber.19901230323.
- ↑ Paquette, L. A.; Wright, J.; Drtina, G. J.; Roberts, R. A. (1987). "Enantiospecific total synthesis of natural (−)-retigeranic acid a and two (−)-retigeranic acid B candidates". J. Org. Chem. 52 (13): 2960. doi:10.1021/jo00389a070.
- ↑ Marshall, J. A.; Lebreton, J. (1988). "Enantioselective synthesis of macrocyclic propargylic alcohols by [2,3] Wittig ring contraction. Synthesis of (+)-aristolactone and cembranoid precursors". J. Am. Chem. Soc. 110 (9): 2925. doi:10.1021/ja00217a039.
- ↑ Castedo, L.; Granja, J. R.; Mourino, A. (1985). "(2,3)-Wittig sigmatropic rearrangements in steroid synthesis. New stereocontrolled approach to steroidal side chains at C-20". Tetrahedron Lett. 26 (40): 4959. doi:10.1016/S0040-4039(00)94997-9.
- ↑ Mikami, K.; Kishi, N.; Nakai, T. (1989). "Silicon-directed regiocontrol in Witting rearrangements of bis-allyl ethers and allyl propargyl ethers". Chem. Lett. (9): 1683. doi:10.1246/cl.1989.1683.
- ↑ Takahashi, O.; Mikami, K.; Nakai, T. (1987). "Asymmetric [2,3]-Wittig rearrangement involving a chiral ester enolate terminus. A chiral synthesis of erythro-.ALPHA.-hydroxy-.BETA.-alkyl carboxylic acid derivatives". Chem. Lett.: 69. doi:10.1246/cl.1987.69.
- ↑ Uemura, M.; Nishimura, H.; Minami, T.; Hayashi, Y. (1991). "(.eta.6-Arene)chromium complexes in organic synthesis: Synthesis of (.+-.)-dihydroxyserrulatic acid". J. Am. Chem. Soc. 113 (14): 5402. doi:10.1021/ja00014a036.
- ↑ Mikami, K.; Nakai, T. (1982). "Applications of the tandem (2,3)-Wittig-oxy-Cope rearrangement to syntheses of exo-brevicomin and oxocrinol. The scope and limitation of the sigmatropic sequences as a synthetic method for δ,ε-unsaturated ketones". Chem. Lett. (9): 1349. doi:10.1246/cl.1982.1349.