Virtual screening
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.[1][2]
Virtual screening has been defined as the "automatically evaluating very large libraries of compounds" using computer programs.[3] As this definition suggests, VS has largely been a numbers game focusing on how the enormous chemical space of over 1060 conceivable compounds[4] can be filtered to a manageable number that can be synthesized, purchased, and tested. Although searching the entire chemical universe may be a theoretically interesting problem, more practical VS scenarios focus on designing and optimizing targeted combinatorial libraries and enriching libraries of available compounds from in-house compound repositories or vendor offerings. As the accuracy of the method has increased, virtual screening has become an integral part of the drug discovery process.[5]
Methods
There are two broad categories of screening techniques: ligand-based and structure-based.[6]
Ligand-based
Given a set of structurally diverse ligands that binds to a receptor, a model of the receptor can be built by exploiting the collective information contained in such set of ligands. These are known as pharmacophore models. A candidate ligand can then be compared to the pharmacophore model to determine whether it is compatible with it and therefore likely to bind.[7]
A different strategy is to develop logic-based rules describing features of substructures and chemical properties related to activity using support vector inductive logic programming.[8] The logic-based features provide insights into activity which can be understood by medicinal chemists. Support vector machine integrate the features to yield a quantitative QSAR, which is then used to screen a database of molecules. This approach is well suited to scaffold hopping to identify novel active molecules and is implemented in the package INDDEx.
Another approach to ligand-based virtual screening is to use 2D chemical similarity analysis methods[9] to scan a database of molecules against one or more active ligand structure.
A popular approach to ligand-based virtual screening is based on searching molecules with shape similar to that of known actives, as such molecules will fit the target's binding site and hence will be likely to bind the target. There are a number of prospective applications of this class of techniques in the literature.[10][11]
Ligand-based virtual screening methods have been extensively compared on the large ChEMBL database in several machine learning challenges, where Deep Learning emerged as the best performing technique.[12][13][14][15][16]
Structure-based
Structure-based virtual screening involves docking of candidate ligands into a protein target followed by applying a scoring function to estimate the likelihood that the ligand will bind to the protein with high affinity.[17][18]
Computing Infrastructure
The computation of pair-wise interactions between atoms, which is a prerequisite for the operation of many virtual screening programs, is of 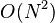 computational complexity, where N is the number of atoms in the system. Because of the quadratic scaling with respect to the number of atoms, the computing infrastructure may vary from a laptop computer for a ligand-based method to a mainframe for a structure-based method.
computational complexity, where N is the number of atoms in the system. Because of the quadratic scaling with respect to the number of atoms, the computing infrastructure may vary from a laptop computer for a ligand-based method to a mainframe for a structure-based method.
Ligand-based
Ligand-based methods typically require a fraction of a second for a single structure comparison operation. A single CPU is enough to perform a large screening within hours. However, several comparisons can be made in parallel in order to expedite the processing of a large database of compounds.
Structure-based
The size of the task requires a parallel computing infrastructure, such as a cluster of Linux systems, running a batch queue processor to handle the work, such as Sun Grid Engine or Torque PBS.
A means of handling the input from large compound libraries is needed. This requires a form of compound database that can be queried by the parallel cluster, delivering compounds in parallel to the various compute nodes. Commercial database engines may be too ponderous, and a high speed indexing engine, such as Berkeley DB, may be a better choice. Furthermore, it may not be efficient to run one comparison per job, because the ramp up time of the cluster nodes could easily outstrip the amount of useful work. To work around this, it is necessary to process batches of compounds in each cluster job, aggregating the results into some kind of log file. A secondary process, to mine the log files and extract high scoring candidates, can then be run after the whole experiment has been run.
Accuracy
The aim of virtual screening is to identify molecules of novel chemical structure that bind to the macromolecular target of interest. Thus, success of a virtual screen is defined in terms of finding interesting new scaffolds rather than the total number of hits. Interpretations of virtual screening accuracy should therefore be considered with caution. Low hit rates of interesting scaffolds are clearly preferable over high hit rates of already known scaffolds.
Most tests of virtual screening studies in the literature are retrospective. In these studies, the performance of a VS technique is measured by its ability to retrieve a small set of previously known molecules with affinity to the target of interest (active molecules or just actives) from a library containing a much higher proportion of assumed inactives or decoys. By contrast, in prospective applications of virtual screening, the resulting hits are subjected to experimental confirmation (e.g., IC50 measurements). There is consensus that retrospective benchmarks are not good predictors of prospective performance and consequently only prospective studies constitute conclusive proof of the suitability of a technique for a particular target.[19][20][21][22]
See also
- Grid computing
- High-throughput screening
- Drug discovery
- Docking (molecular)
- Scoring functions
- ZINC database
References
- ↑ Rester, U (July 2008). "From virtuality to reality - Virtual screening in lead discovery and lead optimization: A medicinal chemistry perspective". Curr Opin Drug Discov Devel 11 (4): 559–68. PMID 18600572.
- ↑ Rollinger JM, Stuppner H, Langer T (2008). "Virtual screening for the discovery of bioactive natural products". Prog Drug Res. Progress in Drug Research 65 (211): 213–49. doi:10.1007/978-3-7643-8117-2_6. ISBN 978-3-7643-8098-4. PMID 18084917.
- ↑ Walters WP, Stahl MT, Murcko MA (1998). "Virtual screening – an overview". Drug Discov. Today 3 (4): 160–178. doi:10.1016/S1359-6446(97)01163-X.
- ↑ Bohacek RS, McMartin C, Guida WC (1996). "The art and practice of structure-based drug design: a molecular modeling perspective". Med. Res. Rev. 16: 3–50. doi:10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6.
- ↑ McGregor, Malcolm J; Luo, Zhaowen; Jiang, Xuliang (June 11, 2007). "Chapter 3: Virtual screening in drug discovery". In Huang, Ziwei. Drug Discovery Research. New Frontiers in the Post-Genomic Era. Wiley-VCH: Weinheim, Germany. pp. 63–88. ISBN 978-0-471-67200-5.
- ↑ McInnes C (2007). "Virtual screening strategies in drug discovery". Curr Opin Chem Biol 11 (5): 494–502. doi:10.1016/j.cbpa.2007.08.033. PMID 17936059.
- ↑ Sun H (2008). "Pharmacophore-based virtual screening". Curr Med Chem 15 (10): 1018–24. doi:10.2174/092986708784049630. PMID 18393859.
- ↑ Reynolds CR, Amini AC, Muggleton SH, Sternberg MJ (2012). "Assessment of a Rule-Based Virtual Screening Technology (INDDEx) on a Benchmark Data Set". J Phys Chem 116 (23): 6732–6739. doi:10.1021/jp212084f.
- ↑ Willet P, Barnard JM, Downs GM (1998). "Chemical similarity searching". J Chem Inf Comput Sci 38 (6): 983–996. doi:10.1021/ci9800211.
- ↑ Rush TS, Grant JA, Mosyak L, Nicholls A (2005). "A Shape-Based 3-D Scaffold Hopping Method and Its Application to a Bacterial Protein−Protein Interaction". Journal of Medicinal Chemistry 48: 1489–1495. doi:10.1021/jm040163o.
- ↑ Ballester PJ, Westwood I, Laurieri N, Sim E, Richards WG (2010). "Prospective virtual screening with Ultrafast Shape Recognition: the identification of novel inhibitors of arylamine N-acetyltransferases". Journal of The Royal Society Interface 7 (43): 335–342. doi:10.1098/rsif.2009.0170. PMC 2842611. PMID 19586957.
- ↑ "Announcement of the winners of the Merck Molecular Activity Challenge" https://www.kaggle.com/c/MerckActivity/details/winners.
- ↑ Dahl, G. E.; Jaitly, N; & Salakhutdinov, R. (2014) "Multi-task Neural Networks for QSAR Predictions," ArXiv, 2014.
- ↑ "Toxicology in the 21st century Data Challenge" https://tripod.nih.gov/tox21/challenge/leaderboard.jsp
- ↑ "NCATS Announces Tox21 Data Challenge Winners" http://www.ncats.nih.gov/news-and-events/features/tox21-challenge-winners.html
- ↑ Unterthiner, T.; Mayr, A.; Klambauer, G.; Steijaert, M.; Ceulemans, H.; Wegner, J. K.; & Hochreiter, S. (2014) "Deep Learning as an Opportunity in Virtual Screening". Workshop on Deep Learning and Representation Learning (NIPS2014).
- ↑ Kroemer RT (2007). "Structure-based drug design: docking and scoring". Curr Protein Pept Sci 8 (4): 312–28. doi:10.2174/138920307781369382. PMID 17696866.
- ↑ Cavasotto CN, Orry AJ (2007). "Ligand docking and structure-based virtual screening in drug discovery". Curr Top Med Chem 7 (10): 1006–14. doi:10.2174/156802607780906753. PMID 17508934.
- ↑ Irwin J (2008). "Community benchmarks for virtual screening". Journal of Computer-Aided Molecular Design 22 (3-4): 193–199. doi:10.1007/s10822-008-9189-4.
- ↑ Good AC, Oprea TI (2008). "Optimization of CAMD techniques 3. Virtual screening enrichment studies: a help or hindrance in tool selection?". Journal of Computer-Aided Molecular Design 22 (3-4): 169–78. doi:10.1007/s10822-007-9167-2.
- ↑ Schneider G (2010). "Virtual screening: an endless staircase?". Nature Reviews Drug Discovery 9: 273–276. doi:10.1038/nrd3139. PMID 20357802.
- ↑ Ballester PJ (2011). "Ultrafast shape recognition: method and applications". Future Medicinal Chemistry 3 (1): 65–78. doi:10.4155/fmc.10.280.
Further reading
- Melagraki G, Afantitis A, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O (2007). "Optimization of biaryl piperidine and 4-amino-2-biarylurea MCH1 receptor antagonists using QSAR modeling, classification techniques and virtual screening". J. Comput. Aided Mol. Des. 21 (5): 251–67. doi:10.1007/s10822-007-9112-4. PMID 17377847.
- Afantitis A, Melagraki G, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O (2006). "Investigation of substituent effect of 1-(3,3-diphenylpropyl)-piperidinyl phenylacetamides on CCR5 binding affinity using QSAR and virtual screening techniques". J. Comput. Aided Mol. Des. 20 (2): 83–95. doi:10.1007/s10822-006-9038-2. PMID 16783600.
- Eckert H, Bajorath J (2007). "Molecular similarity analysis in virtual screening: foundations, limitations and novel approaches". Drug Discov. Today 12 (5–6): 225–33. doi:10.1016/j.drudis.2007.01.011. PMID 17331887.
- Willett P (2006). "Similarity-based virtual screening using 2D fingerprints". Drug Discov. Today 11 (23–24): 1046–53. doi:10.1016/j.drudis.2006.10.005. PMID 17129822.
- Fara DC, Oprea TI, Prossnitz ER, Bologa CG, Edwards BS, Sklar LA (2006). "Integration of virtual and physical screening". Drug Discov. Today: Technologies 3 (4): 377–385. doi:10.1016/j.ddtec.2006.11.003.
- Muegge I, Oloffa S (2006). "Advances in virtual screening". Drug Discov. Today: Technologies 3 (4): 405–411. doi:10.1016/j.ddtec.2006.12.002.
- Schneider G (April 2010). "Virtual screening: an endless staircase?". Nat Rev Drug Discov 9 (4): 273–6. doi:10.1038/nrd3139. PMID 20357802.
External links
- MolSoft ICM — ligand and structure-based virtual screening
- Glide — ligand-receptor docking and virtual screening software
- INDDEx — ligand-based hit discovery for scaffold hopping using SVILP machine learning
- ZINC — a free database of commercially-available compounds for virtual screening.
- Virtual Screening Methods
- mcule — Easy-to-use, online virtual screening.
- Free service to screen for GPCR ligands, ion channel blockers and kinase inhibitors
- Brutus — a similarity analysis tool for ligand-based virtual screening.
- NovaMechanics Cheminformatics Research Combined structure & ligand based chemistry driven virtual screening.