Tyrosinemia type III
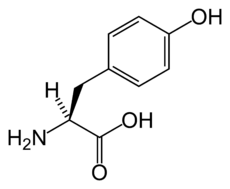
Type III tyrosinemia is a rare disorder caused by a deficiency of the enzyme 4-hydroxyphenylpyruvate dioxygenase (EC 1.13.11.27), encoded by the gene HPD. This enzyme is abundant in the liver, and smaller amounts are found in the kidneys. It is one of a series of enzymes needed to break down tyrosine. Specifically, 4-hydroxyphenylpyruvate dioxygenase converts a tyrosine byproduct called 4-hydroxyphenylpyruvate to homogentisic acid. Characteristic features of type III tyrosinemia include mild mental retardation, seizures, and periodic loss of balance and coordination (intermittent ataxia). Type III tyrosinemia is very rare; only a few cases have been reported.

Pathophysiology of metabolic disorders of tyrosine, resulting in elevated levels of tyrosine in blood.
|
|---|
| | K→acetyl-CoA | |
|---|
| | G | |
|---|
| Transport/
IE of RTT | |
|---|
| | Other | |
|---|
| |
|---|
| | Description |
- Metabolism
- Enzymes and pathways: citric acid cycle
- pentose phosphate
- glycoproteins
- glycosaminoglycans
- phospholipid
- cholesterol and steroid
- sphingolipids
- eicosanoids
- amino acid
- urea cycle
- nucleotide
|
|---|
| | Disorders |
- Citric acid cycle and electron transport chain
- Glycoprotein
- Proteoglycan
- Fatty-acid
- Phospholipid
- Cholesterol and steroid
- Eicosanoid
- Amino acid
- Purine-pyrimidine
- Heme metabolism
- Symptoms and signs
|
|---|
| | Treatment | |
|---|
|
|