Thermodynamic potential
A thermodynamic potential is a scalar quantity used to represent the thermodynamic state of a system. The concept of thermodynamic potentials was introduced by Pierre Duhem in 1886. Josiah Willard Gibbs in his papers used the term fundamental functions. One main thermodynamic potential that has a physical interpretation is the internal energy U. It is the energy of configuration of a given system of conservative forces (that is why it is a potential) and only has meaning with respect to a defined set of references (or data). Expressions for all other thermodynamic energy potentials are derivable via Legendre transforms from an expression for U. In thermodynamics, certain forces, such as gravity, are typically disregarded when formulating expressions for potentials. For example, while all the working fluid in a steam engine may have higher energy due to gravity while sitting on top of Mount Everest than it would at the bottom of the Mariana Trench, the gravitational potential energy term in the formula for the internal energy would usually be ignored because changes in gravitational potential within the engine during operation would be negligible.
| Thermodynamics | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
 The classical Carnot heat engine | ||||||||||||
|
Branches |
||||||||||||
|
||||||||||||
| Book:Thermodynamics | ||||||||||||
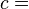
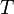

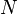


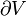



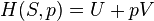
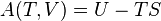
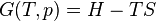
Description and interpretation
Five common thermodynamic potentials are:[1]
| Name | Symbol | Formula | Natural variables |
|---|---|---|---|
| Internal energy |  |
 |
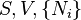 |
| Helmholtz free energy | 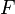 |
 |
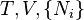 |
| Enthalpy | 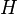 |
 |
 |
| Gibbs free energy | 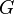 |
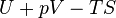 |
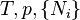 |
| Landau Potential (Grand potential) | 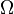 , , 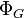 |
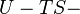 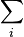  |
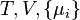 |
where T = temperature, S = entropy, p = pressure, V = volume. The Helmholtz free energy is often denoted by the symbol F, but the use of A is preferred by IUPAC,[2] ISO and IEC.[3] Ni is the number of particles of type i in the system and μi is the chemical potential for an i-type particle. For the sake of completeness, the set of all Ni are also included as natural variables, although they are sometimes ignored.
These five common potentials are all energy potentials, but there are also entropy potentials. The thermodynamic square can be used as a tool to recall and derive some of the potentials.
Just as in mechanics, where potential energy is defined as capacity to do work, similarly different potentials have different meanings. Internal energy (U ) is the capacity to do work plus the capacity to release heat. Gibbs energy is the capacity to do non-mechanical work. Enthalpy is the capacity to do non-mechanical work plus the capacity to release heat. Helmholtz free energy is the capacity to do mechanical work (useful work). From these definitions we can say that ΔU is the energy added to the system, ΔF is the total work done on it, ΔG is the non-mechanical work done on it, and ΔH is the sum of non-mechanical work done on the system and the heat given to it. Thermodynamic potentials are very useful when calculating the equilibrium results of a chemical reaction, or when measuring the properties of materials in a chemical reaction. The chemical reactions usually take place under some simple constraints such as constant pressure and temperature, or constant entropy and volume, and when this is true, there is a corresponding thermodynamic potential that comes into play. Just as in mechanics, the system will tend towards lower values of potential and at equilibrium, under these constraints, the potential will take on an unchanging minimum value. The thermodynamic potentials can also be used to estimate the total amount of energy available from a thermodynamic system under the appropriate constraint.
In particular: (see principle of minimum energy for a derivation)[4]
- When the entropy (S ) and "external parameters" (e.g. volume) of a closed system are held constant, the internal energy (U ) decreases and reaches a minimum value at equilibrium. This follows from the first and second laws of thermodynamics and is called the principle of minimum energy. The following three statements are directly derivable from this principle.
- When the temperature (T ) and external parameters of a closed system are held constant, the Helmholtz free energy (F ) decreases and reaches a minimum value at equilibrium.
- When the pressure (p) and external parameters of a closed system are held constant, the enthalpy (H ) decreases and reaches a minimum value at equilibrium.
- When the temperature (T ), pressure (p) and external parameters of a closed system are held constant, the Gibbs free energy (G ) decreases and reaches a minimum value at equilibrium.
Natural variables
The variables that are held constant in this process are termed the natural variables of that potential.[5] The natural variables are important not only for the above-mentioned reason, but also because if a thermodynamic potential can be determined as a function of its natural variables, all of the thermodynamic properties of the system can be found by taking partial derivatives of that potential with respect to its natural variables and this is true for no other combination of variables. On the converse, if a thermodynamic potential is not given as a function of its natural variables, it will not, in general, yield all of the thermodynamic properties of the system.
Notice that the set of natural variables for the above four potentials are formed from every combination of the T-S and P-V variables, excluding any pairs of conjugate variables. There is no reason to ignore the Ni − μi conjugate pairs, and in fact we may define four additional potentials for each species.[6] Using IUPAC notation in which the brackets contain the natural variables (other than the main four), we have:
| Formula | Natural variables |
![U[\mu_j]=U-\mu_jN_j\,](../I/m/269912f4aa53f453130defff4762ac97.png) |
 |
![F[\mu_j]=U-TS-\mu_jN_j\,](../I/m/ae0ec452436604d9a814792e19e1dbef.png) |
 |
![H[\mu_j]=U+pV-\mu_jN_j\,](../I/m/490ddd5ef91f255a4acf049d99b48e24.png) |
 |
![G[\mu_j]=U+pV-TS-\mu_jN_j\,](../I/m/2abfd370faa7bb1e364a14fb58c493f3.png) |
 |
If there is only one species, then we are done. But, if there are, say, two species, then there will be additional potentials such as ![U[\mu_1,\mu_2] = U-\mu_1 N_1-\mu_2 N_2](../I/m/f224c9a2320a5e73eaa99a56a805ffd7.png) and so on. If there are D dimensions to the thermodynamic space, then there are 2D unique thermodynamic potentials. For the most simple case, a single phase ideal gas, there will be three dimensions, yielding eight thermodynamic potentials.
and so on. If there are D dimensions to the thermodynamic space, then there are 2D unique thermodynamic potentials. For the most simple case, a single phase ideal gas, there will be three dimensions, yielding eight thermodynamic potentials.
The fundamental equations
The definitions of the thermodynamic potentials may be differentiated and, along with the first and second laws of thermodynamics, a set of differential equations known as the fundamental equations follow.[7] (Actually they are all expressions of the same fundamental thermodynamic relation, but are expressed in different variables.) By the first law of thermodynamics, any differential change in the internal energy U of a system can be written as the sum of heat flowing into the system and work done by the system on the environment, along with any change due to the addition of new particles to the system:

where δQ is the infinitesimal heat flow into the system, and δW is the infinitesimal work done by the system, μi is the chemical potential of particle type i and Ni is the number of type i particles. (Note that neither δQ nor δW are exact differentials. Small changes in these variables are, therefore, represented with δ rather than d.)
By the second law of thermodynamics, we can express the internal energy change in terms of state functions and their differentials. In case of reversible changes we have:


where
- T is temperature,
- S is entropy,
- p is pressure,
and V is volume, and the equality holds for reversible processes.
This leads to the standard differential form of the internal energy in case of a quasistatic reversible change:

Since U, S and V are thermodynamic functions of state, the above relation holds also for arbitrary non-reversible changes. If the system has more external variables than just the volume that can change, the fundamental thermodynamic relation generalizes to:

Here the Xi are the generalized forces corresponding to the external variables xi.
Applying Legendre transforms repeatedly, the following differential relations hold for the four potentials:
 |  |  |
 |  |  | |
 | | |  |
| | |
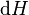 | | 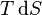 |
 |  | | |
 | | | |
| | |
Note that the infinitesimals on the right-hand side of each of the above equations are of the natural variables of the potential on the left-hand side. Similar equations can be developed for all of the other thermodynamic potentials of the system. There will be one fundamental equation for each thermodynamic potential, resulting in a total of 2D fundamental equations.
The differences between the four thermodynamic potentials can be summarized as follows:


The equations of state
We can use the above equations to derive some differential definitions of some thermodynamic parameters. If we define Φ to stand for any of the thermodynamic potentials, then the above equations are of the form:

where xi and yi are conjugate pairs, and the yi are the natural variables of the potential Φ. From the chain rule it follows that:

Where yi ≠ j is the set of all natural variables of Φ except yi . This yields expressions for various thermodynamic parameters in terms of the derivatives of the potentials with respect to their natural variables. These equations are known as equations of state since they specify parameters of the thermodynamic state.[8] If we restrict ourselves to the potentials U, F, H and G, then we have:




where, in the last equation, ϕ is any of the thermodynamic potentials U, F, H, G and  are the set of natural variables for that potential, excluding Ni . If we use all potentials, then we will have more equations of state such as
are the set of natural variables for that potential, excluding Ni . If we use all potentials, then we will have more equations of state such as
![-N_j=\left(\frac{\partial U[\mu_j]}{\partial \mu_j}\right)_{S,V,\{N_{i\ne j}\}}](../I/m/3c18bf0022d4ea560cad992fb8ec60e5.png)
and so on. In all, there will be D equations for each potential, resulting in a total of D 2D equations of state. If the D equations of state for a particular potential are known, then the fundamental equation for that potential can be determined. This means that all thermodynamic information about the system will be known, and that the fundamental equations for any other potential can be found, along with the corresponding equations of state.
The Maxwell relations
Again, define xi and yi to be conjugate pairs, and the yi to be the natural variables of some potential Φ. We may take the "cross differentials" of the state equations, which obey the following relationship:

From these we get the Maxwell relations.[1][9] There will be (D − 1)/2 of them for each potential giving a total of D(D − 1)/2 equations in all. If we restrict ourselves the U, F, H, G



Using the equations of state involving the chemical potential we get equations such as:

and using the other potentials we can get equations such as:
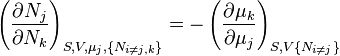
Euler integrals
Again, define xi and yi to be conjugate pairs, and the yi to be the natural variables of the internal energy. Since all of the natural variables of the internal energy U are extensive quantities

it follows from Euler's homogeneous function theorem that the internal energy can be written as:

From the equations of state, we then have:
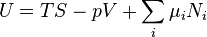
Substituting into the expressions for the other main potentials we have:

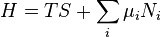
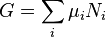
As in the above sections, this process can be carried out on all of the other thermodynamic potentials. Note that the Euler integrals are sometimes also referred to as fundamental equations.
The Gibbs–Duhem relation
Deriving the Gibbs–Duhem equation from basic thermodynamic state equations is straightforward.[7][10][11] Equating any thermodynamic potential definition with its Euler integral expression yields:

Differentiating, and using the second law:
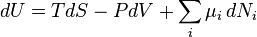
yields:

Which is the Gibbs–Duhem relation. The Gibbs–Duhem is a relationship among the intensive parameters of the system. It follows that for a simple system with I components, there will be I + 1 independent parameters, or degrees of freedom. For example, a simple system with a single component will have two degrees of freedom, and may be specified by only two parameters, such as pressure and volume for example. The law is named after Josiah Willard Gibbs and Pierre Duhem.
Chemical reactions
Changes in these quantities are useful for assessing the degree to which a chemical reaction will proceed. The relevant quantity depends on the reaction conditions, as shown in the following table. Δ denotes the change in the potential and at equilibrium the change will be zero.
| Constant V | Constant p | |
|---|---|---|
| Constant S | ΔU | ΔH |
| Constant T | ΔF | ΔG |
Most commonly one considers reactions at constant p and T, so the Gibbs free energy is the most useful potential in studies of chemical reactions.
See also
Notes
References
- Alberty, R. A. (2001). "Use of Legendre transforms in chemical thermodynamics" (PDF). Pure Appl. Chem. 73 (8): 1349–1380. doi:10.1351/pac200173081349.
- Callen, Herbert B. (1985). Thermodynamics and an Introduction to Themostatistics (2nd ed.). New York: John Wiley & Sons. ISBN 0-471-86256-8.
- Moran, Michael J.; Shapiro, Howard N. (1996). Fundamentals of Engineering Thermodynamics (3rd ed.). New York ; Toronto: J. Wiley & Sons. ISBN 0-471-07681-3.
Further reading
- McGraw Hill Encyclopaedia of Physics (2nd Edition), C.B. Parker, 1994, ISBN 0-07-051400-3
- Thermodynamics, From Concepts to Applications (2nd Edition), A. Shavit, C. Gutfinger, CRC Press (Taylor and Francis Group, USA), 2009, ISBN 9781420073683
- Chemical Thermodynamics, D.J.G. Ives, University Chemistry, Macdonald Technical and Scientific, 1971, ISBN 0-356-03736-3
- Elements of Statistical Thermodynamics (2nd Edition), L.K. Nash, Principles of Chemistry, Addison-Wesley, 1974, ISBN 0-201-05229-6
- Statistical Physics (2nd Edition), F. Mandl, Manchester Physics, John Wiley & Sons, 2008, ISBN 9780471566588
External links
- Thermodynamic Potentials – Georgia State University
- Chemical Potential Energy: The 'Characteristic' vs the Concentration-Dependent Kind