Synaptic plasticity
In neuroscience, synaptic plasticity is the ability of synapses to strengthen or weaken over time, in response to increases or decreases in their activity.[1] Plastic change also results from the alteration of the number of neurotransmitter receptors located on a synapse.[2] There are several underlying mechanisms that cooperate to achieve synaptic plasticity, including changes in the quantity of neurotransmitters released into a synapse and changes in how effectively cells respond to those neurotransmitters.[3] Synaptic plasticity in both excitatory and inhibitory synapses has been found to be dependent upon postsynaptic calcium release.[2] Since memories are postulated to be represented by vastly interconnected networks of synapses in the brain, synaptic plasticity is one of the important neurochemical foundations of learning and memory (see Hebbian theory).
Historical discoveries
In 1973, Terje Lømo and Tim Bliss first described the now widely studied phenomenon of long-term potentiation (LTP) in a publication in the Journal of Physiology. The experiment described was conducted on the synapse between the perforant path and dentate gyrus in the hippocampi of anaesthetised rabbits. They were able to show a burst of tetanic (100 Hz) stimulus on perforant path fibres led to a dramatic and long-lasting augmentation in the post-synaptic response of cells onto which these fibres synapse in the dentate gyrus. In the same year, the pair published very similar data recorded from awake rabbits. This discovery was of particular interest due to the proposed role of the hippocampus in certain forms of memory.
Biochemical mechanisms
Two molecular mechanisms for synaptic plasticity (researched by the Eric Kandel laboratories) involve the NMDA and AMPA glutamate receptors. Opening of NMDA channels (which relates to the level of cellular depolarization) leads to a rise in post-synaptic Ca2+ concentration and this has been linked to long-term potentiation, LTP (as well as to protein kinase activation); strong depolarization of the post-synaptic cell completely displaces the magnesium ions that block NMDA ion channels and allows calcium ions to enter a cell – probably causing LTP, while weaker depolarization only partially displaces the Mg2+ ions, resulting in less Ca2+ entering the post-synaptic neuron and lower intracellular Ca2+ concentrations (which activate protein phosphatases and induce long-term depression, LTD).[4]
These activated protein kinases serve to phosphorylate post-synaptic excitatory receptors (e.g. AMPA receptors), improving cation conduction, and thereby potentiating the synapse. Also, these signals recruit additional receptors into the post-synaptic membrane, stimulating the production of a modified receptor type, thereby facilitating an influx of calcium. This in turn increases post-synaptic excitation by a given pre-synaptic stimulus. This process can be reversed via the activity of protein phosphatases, which act to dephosphorylate these cation channels.[5]
The second mechanism depends on a second messenger cascade regulating gene transcription and changes in the levels of key proteins at synapses such as CaMKII and PKAII. Activation of the second messenger pathway leads to increased levels of CaMKII and PKAII within the dendritic spine. These protein kinases have been linked to growth in dendritic spine volume and LTP processes such as the addition of AMPA receptors to the plasma membrane and phosphorylation of ion channels for enhanced permeability.[6] Localization or compartmentalization of activated proteins occurs in the presence of their given stimulus which creates local effects in the dendritic spine. Calcium influx from NMDA receptors is necessary for the activation of CaMKII. This activation is localized to spines with focal stimulation and is inactivated before spreading to adjacent spines or the shaft, indicating an important mechanism of LTP in that particular changes in protein activation can be localized or compartmentalized to enhance the responsivity of single dendritic spines. Individual dendritic spines are capable of forming unique responses to presynaptic cells.[7] This second mechanism can be triggered by protein phosphorylation but takes longer and lasts longer, providing the mechanism for long-lasting memory storage. The duration of the LTP can be regulated by breakdown of these second messengers. Phosphodiesterase, for example, breaks down the secondary messenger cAMP, which has been implicated in increased AMPA receptor synthesis in the post-synaptic neuron .
Long-lasting changes in the efficacy of synaptic connections (long-term potentiation, or LTP) between two neurons can involve the making and breaking of synaptic contacts. Genes such as activin ß-A, which encodes a subunit of activin A, are up-regulated during early stage LTP. The activin molecule modulates the actin dynamics in dendritic spines through the MAP kinase pathway. By changing the F-actin cytoskeletal structure of dendritic spines, spines are lengthened and the chance that they make synaptic contacts with the axonal terminals of the presynaptic cell is increased. The end result is long-term maintenance of LTP.[8]
The number of ion channels on the post-synaptic membrane affects the strength of the synapse.[9] Research suggests that the density of receptors on post-synaptic membranes changes, affecting the neuron’s excitability in response to stimuli. In a dynamic process that is maintained in equilibrium, N-methyl D-aspartate receptor (NMDA receptor) and AMPA receptors are added to the membrane by exocytosis and removed by endocytosis.[10][11][12] These processes, and by extension the number of receptors on the membrane, can be altered by synaptic activity.[10][12] Experiments have shown that AMPA receptors are delivered to the synapse through vesicular membrane fusion with the postsynaptic membrane via the protein kinase CaMKII, which is activated by the influx of calcium through NMDA receptors. CaMKII also improves AMPA ionic conductance through phosphorylation.[13] When there is high-frequency NMDA receptor activation, there is an increase in the expression of a protein PSD-95 that increases synaptic capacity for AMPA receptors.[14] This is what leads to a long-term increase in AMPA receptors and thus synaptic strength and plasticity.
If the strength of a synapse is only reinforced by stimulation or weakened by its lack, a positive feedback loop will develop, causing some cells never to fire and some to fire too much. But two regulatory forms of plasticity, called scaling and metaplasticity, also exist to provide negative feedback.[12] Synaptic scaling is a primary mechanism by which a neuron is able to stabilize firing rates up or down.[15]
Synaptic scaling serves to maintain the strengths of synapses relative to each other, lowering amplitudes of small excitatory postsynaptic potentials in response to continual excitation and raising them after prolonged blockage or inhibition.[12] This effect occurs gradually over hours or days, by changing the numbers of NMDA receptors at the synapse (Pérez-Otaño and Ehlers, 2005). Metaplasticity varies the threshold level at which plasticity occurs, allowing integrated responses to synaptic activity spaced over time and preventing saturated states of LTP and LTD. Since LTP and LTD (long-term depression) rely on the influx of Ca2+ through NMDA channels, metaplasticity may be due to changes in NMDA receptors, altered calcium buffering, altered states of kinases or phosphatases and a priming of protein synthesis machinery.[16] Synaptic scaling is a primary mechanism by which a neuron to be selective to its varying inputs.[17] The neuronal circuitry affected by LTP/LTD and modified by scaling and metaplasticity leads to reverberatory neural circuit development and regulation in a Hebbian manner which is manifested as memory, whereas the changes in neural circuitry, which begin at the level of the synapse, are an integral part in the ability of an organism to learn.[18]
There is also a specificity element of biochemical interactions to create synaptic plasticity, namely the importance of location. Processes occur at microdomains – such as exocytosis of AMPA receptors is spatially regulated by the t-SNARE STX4.[19] Specificity is also an important aspect of CAMKII signaling involving nanodomain calcium.[20] The spatial gradient of PKA between dendritic spines and shafts is also important for the strength and regulation of synaptic plasticity.[6] It is important to remember that the biochemical mechanisms altering synaptic plasticity occur at the level of individual synapses of a neuron. Since the biochemical mechanisms are confined to these "microdomains," the resulting synaptic plasticity affects only the specific synapse at which it took place.
Theoretical mechanisms
A bidirectional model, describing both LTP and LTD, of synaptic plasticity has proved necessary for a number of different learning mechanisms in computational neuroscience, neural networks, and biophysics. Three major hypotheses for the molecular nature of this plasticity have been well-studied, and none are required to be the exclusive mechanism:
- Change in the probability of glutamate release.
- Insertion or removal of post-synaptic AMPA receptors.
- Phosphorylation and de-phosphorylation inducing a change in AMPA receptor conductance.
Of these, the first two hypotheses have been recently mathematically examined to have identical calcium-dependent dynamics which provides strong theoretical evidence for a calcium-based model of plasticity, which in a linear model where the total number of receptors are conserved looks like
![\frac{d W_i(t)}{d t}=\frac{1}{\tau([Ca^{2+}]_i)}\left(\Omega([Ca^{2+}]_i)-W_i\right),](../I/m/3d351dfdab0857ce70a53efbc41ca672.png)
where 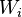 is the synaptic weight of the
is the synaptic weight of the 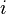 th input axon,
th input axon, 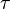 is a time constant dependent on the insertion and removal rates of neurotransmitter receptors, which is dependent on
is a time constant dependent on the insertion and removal rates of neurotransmitter receptors, which is dependent on ![[Ca^{2+}]](../I/m/6dd9714258be1b1c0423ad232f93bf25.png) , the concentration of calcium.
, the concentration of calcium.  is also a function of the concentration of calcium that depends linearly on the number of receptors on the membrane of the neuron at some fixed point. Both
is also a function of the concentration of calcium that depends linearly on the number of receptors on the membrane of the neuron at some fixed point. Both 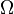 and are found experimentally and agree on results from both hypotheses. The model makes important simplifications that make it unsuited for actual experimental predictions, but provides a significant basis for the hypothesis of a calcium-based synaptic plasticity dependence.[21]
and are found experimentally and agree on results from both hypotheses. The model makes important simplifications that make it unsuited for actual experimental predictions, but provides a significant basis for the hypothesis of a calcium-based synaptic plasticity dependence.[21]
Short-term plasticity
Short-term synaptic plasticity acts on a timescale of tens of milliseconds to a few minutes unlike long-term plasticity, which lasts from minutes to hours. Short term plasticity can either strengthen or weaken a synapse.
Synaptic enhancement
Short-term synaptic enhancement results from an increased probability of synaptic terminals releasing transmitters in response to pre-synaptic action potentials. Synapses will strengthen for a short time because of either an increase in size of the readily releasable pool of packaged transmitter or an increase in the amount of packaged transmitter released in response to each action potential.[22] Depending on the time scales over which it acts synaptic enhancement is classified as neural facilitation, synaptic augmentation or post-tetanic potentiation.
Synaptic depression
Synaptic fatigue or depression is usually attributed to the depletion of the readily releasable vesicles. Depression can also arise from post-synaptic processes and from feedback activation of presynaptic receptors.[23] Heterosynaptic depression is thought to be linked to the release of adenosine triphosphate (ATP) from astrocytes.[24]
Long-term plasticity
Long-term depression (LTD) and long-term potentiation (LTP) are two forms of long-term plasticity, lasting minutes or more, that occur at excitatory synapses.[2] NMDA-dependent LTD and LTP have been extensively researched, and are found to require the binding of glutamate, and glycine or D-serine for activation of NMDA receptors.[24] The turning point for the synaptic modification of a synapse has been found to be modifiable itself, depending on the history of the synapse.[25] Recently, a number of attempts have been made to offer a comprehensive model that could account for most forms of synaptic plasticity [26]
Long-term depression
Brief activation of an excitatory pathway can produce what is known as long-term depression (LTD) of synaptic transmission in many areas of the brain. LTD is induced by a minimum level of postsynaptic depolarization and simultaneous increase in the intracellular calcium concentration at the postsynaptic neuron. LTD can be initiated at inactive synapses if the calcium concentration is raised to the minimum required level by heterosynaptic activation, or if the extracellular concentration is raised. These alternative conditions capable of causing LTD differ from the Hebb rule, and instead depend on synaptic activity modifications. D-serine release by astrocytes has been found to lead to a significant reduction of LTD in the hippocampus.[24] A LTD was evidenced in 2011 for the electrical synapses (modification of Gap Junctions efficacy through their activity).[27]
Long-term potentiation
Long-term potentiation, commonly referred to as LTP, is an increase in synaptic response following potentiating pulses of electrical stimuli that sustains at a level above the baseline response for hours or longer. LTP involves interactions between postsynaptic neurons and the specific presynaptic inputs that form a synaptic association, and is specific to the stimulated pathway of synaptic transmission. The long-term stabilization of synaptic changes is determined by a parallel increase of pre- and postsynaptic structures such as axonal bouton, dendritic spine and postsynaptic density.[14] On the molecular level, an increase of the postsynaptic scaffolding proteins PSD-95 and Homer1c has been shown to correlate with the stabilization of synaptic enlargement.[14]
Modification of astrocyte coverage at the synapses in the hippocampus has been found to result from the induction of LTP, which has been found to be linked to the release of D-serine, nitric oxide, and the chemokine, s100B by astrocytes.[24] LTP is also a model for studying the synaptic basis of Hebbian plasticity. Induction conditions resemble those described for the initiation of long-term depression (LTD), but a stronger depolarization and a greater increase of calcium are necessary to achieve LTP.[28]
Synaptic strength
The modification of synaptic strength is referred to as functional plasticity. Changes in synaptic strength involve distinct mechanisms of particular types of glial cells, the most researched type being astrocytes.[24]
See also
- BCM theory
- Excitatory postsynaptic potential
- Homosynaptic Plasticity
- Heterosynaptic Plasticity
- Homeostatic plasticity
- Inhibitory postsynaptic potential
- Long-term potentiation (LTP)
- Long-term depression (LTD)
- Activity-dependent plasticity
- Spike-timing-dependent plasticity (STDP)
- Synaptic augmentation (Short-term plasticity)
- Neural facilitation (Short-term plasticity)
- Neuroplasticity
- Postsynaptic potential
- Non-synaptic plasticity
References
- ↑ Hughes, John R. (1958). "Post-tetanic Potentiation". Physiological Reviews 38 (1): 91–113. PMID 13505117.
- ↑ 2.0 2.1 2.2 Gerrow, Kimberly; Antoine (2010). "Synaptic stability and plasticity in a floating world". Current Opinion in Neurobiology 20 (5): 631–639. doi:10.1016/j.conb.2010.06.010. PMID 20655734.
- ↑ Gaiarsa, J.L.; Caillard O., and Ben-Ari Y. (2002). "Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance". Trends in Neurosciences 25 (11): 564–570. doi:10.1016/S0166-2236(02)02269-5. PMID 12392931.
- ↑ Bear MF, Connors BW, and Paradisio MA. 2007. Neuroscience: Exploring the Brain, 3rd ed. Lippincott, Williams & Wilkins
- ↑ Soderling, TR; Derkach, VA (2000). "Postsynaptic protein phosphorylation and LTP". Trends in Neurosciences 23 (2): 75–80. doi:10.1016/S0166-2236(99)01490-3. PMID 10652548.
- ↑ 6.0 6.1 Haining, Z.; Sia G.; Sato T.; Gray N.; Mao T.; Khuchia Z.; Huganir R.; Svodoba K. (2009). "Subcellular Dynamics of Type II PKA in Neurons". Cell Press 62: 363–374. doi:10.1016/j.neuron.2009.03.013.
- ↑ Seok-Jin, R.; Escobedo-Lozoya Y.; Szatmari E.; Yasuda R. (2009). "Activation of CaMKII in single dendritic spines during long-term potentiation". Nature 458 (19): 299–306. doi:10.1038/nature07842. PMC 2719773. PMID 19295602.
- ↑ Shoji-Kasai, Yoko; Hiroshi Ageta; Yoshihisa Hasegawa; Kunihiro Tsuchida; Hiromu Sugino; Kaoru Inokuchi (2007). "Activin increases the number of synaptic contacts and the length of dendritic spine necks by modulating spinal actin dynamics". Journal of Cell Science 120 (Pt 21): 3830–3837. doi:10.1242/jcs.012450. PMID 17940062.
- ↑ Debanne, D.; Daoudal G.; Sourdet V.; Russier M. (2003). "Brain plasticity and ion channels". Journal of Physiology, Paris 97 (4-6): 403–414. doi:10.1016/j.jphysparis.2004.01.004. PMID 15242652.
- ↑ 10.0 10.1 Shi, S.H.; Hayashi Y.; Petralia R.S.; Zaman S.H.; Wenthold R.; Svoboda K.; Malinow R. (1999). "Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation". Science 284 (5421): 1811–1816. doi:10.1126/science.284.5421.1811. ISSN 0193-4511. PMID 10364548.
- ↑ Song, I.; Huganir R.L. (2002). "Regulation of AMPA receptors during synaptic plasticity". Trends in Neurosciences 25 (11): 578–589. doi:10.1016/S0166-2236(02)02270-1. PMID 12392933.
- ↑ 12.0 12.1 12.2 12.3 Pérez-Otaño, I.; Ehlers M.D. (2005). "Homeostatic plasticity and NMDA receptor trafficking" (PDF). Trends in Neurosciences 28 (5): 229–238. doi:10.1016/j.tins.2005.03.004. PMID 15866197. Retrieved 2007-06-08.
- ↑ Bear, M.F. (2007). Neuroscience: Exploring the Brain. Third Edition. Lippincott Williams & Wilkins. p. 779. ISBN 978-0-7817-6003-4.
- ↑ 14.0 14.1 14.2 Meyer, D.; Bonhoeffer T., and Scheuss V. (2014). "Balance and Stability of Synaptic Structures during Synaptic Plasticity". Neuron 82 (2): 430–443. doi:10.1016/j.neuron.2014.02.031. PMID 24742464.
- ↑ Desai, Niraj S.; Robert H. Cudmore, Sacha B. Nelson & Gina G. Turrigiano (2002). "Critical periods for experience-dependent synaptic scaling in visual cortex". Nature Neuroscience 5 (8): 783–789. doi:10.1038/nn878. PMID 12080341.
- ↑ Abraham, Wickliffe; Warren P. Tate (1997). "Metaplasticity: A new vista across the field of synaptic plasticity". Progress in Neurobiology 52 (4): 303–323. doi:10.1016/S0301-0082(97)00018-X. PMID 9247968.
- ↑ Abbott, L.; Sacha B. Nelson (2000). "Synaptic plasticity: taming the beast". Nature Neuroscience 3: 1178–1183. doi:10.1038/81453. PMID 11127835.
- ↑ Cooper, Stephen J. (2005). "Donald O. Hebb's synapse and learning rule: a history and commentary". Neuroscience and Biobehavioral Reviews 28 (8): 851–874. doi:10.1016/j.neubiorev.2004.09.009. PMID 15642626.
- ↑ Kennedy, Matthew J.; Ian G. Davison; Camenzind G. Robinson; Michael D. Ehlers (2010). "Syntaxin-4 Defines a Domain for Activity-Dependent Exocytosis in Dendritic Spines". Cell (Elsevier Inc.) 141 (3): 1–12. doi:10.1016/j.cell.2010.02.042. PMC 2874581. PMID 20434989.
- ↑ Lee, Seok-Jin R.; Yasmin Escobedo-Lozoya; Erzsebet M. Szatmari; Ryohei Yasuda (2009). "Activation of CaMKII in single dendritic spines during long-term potentiation". Nature (Macmillan Publishers Limited) 458 (7236): 299–304. doi:10.1038/nature07842. PMC 2719773. PMID 19295602.
- ↑ Shouval, Harel Z.; Gastone C. Castellani, Brian S. Blais, Luk C. Yeung, Leon N. Cooper (2002). "Converging evidence for a simplified biophysical model of synaptic plasticity". Biological Cybernetics 87 (5-6): 383–391. doi:10.1007/s00422-002-0362-x. PMID 12461628. Retrieved 2007-11-12.
- ↑ Stevens, C. F.; Wesseling, J. F. (1999). "Augmentation is a Potentiation of the Exocytotic Process". Neuron 22 (1): 139–146. doi:10.1016/S0896-6273(00)80685-6. PMID 10027296.
- ↑ Zucker, Robert S.; Regehr, WG (Mar 2002). "Short-term Synaptic Plasticity". Annual Review of Physiology 64: 355–405. doi:10.1146/annurev.physiol.64.092501.114547. PMID 11826273. Retrieved 2010-11-27.
- ↑ 24.0 24.1 24.2 24.3 24.4 Achour, S. Ben; O. Pascaul (Mar 2010). "Glia: The many ways to modulate synaptic plasticity". Neurochemistry International 57 (4): 440–445. doi:10.1016/j.neuint.2010.02.013. PMID 20193723.
- ↑ Bear MF. (1995). Mechanism for a sliding synaptic modification threshold. Neuron, 15(1):1-4
- ↑ Michmizos, D. et al.(2011). Synaptic plasticity: a Unifying Model to address some persisting questions. International Journal of Neuroscience, 121(6): 289-304.
- ↑ J. S. Haas, B. Zavala, C. E. Landisman, Activity-dependent long-term depression of electrical synapses" Science 334, 389–393 (2011). [Abstract] [Full Text]
- ↑ Artola, Alain; Wolf Singer (1993). "Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation". Trends in Neuroscience 16 (11): 480–487. doi:10.1016/0166-2236(93)90081-V. Retrieved 2010-11-28.
Bibliography
- Thornton, James K. (2003). "New LSD Research: Gene Expression within the Mammalian Brain". MAPS 13 (1). Retrieved 2007-06-08.
- Chapouthier, G. (2004). From the search for a molecular code of memory to the role of neurotransmitters: a historical perspective, Neural Plasticity, 11(3-4), 151-158
- Hawkins, R.D., Kandel, E.R., & Bailey, C.H. (June 2006). Molecular Mechanisms of Memory Storage in Aplysia. Biological Bulletin, 210, 174-191.
- LeDoux, Joseph. Synaptic Self: How Our Brains Become Who We Are. New York: Penguin Books, 2002. 1-324. Print.
External links
- Overview
- Finnerty lab, MRC Centre for Neurodegeneration Research, London
- Brain Basics Synaptic Plasticity Synaptic transmission is plastic
- Synaptic Plasticity, Neuroscience Online (electronic neuroscience textbook by UT Houston Medical School)
Videos, podcasts
- Synaptic plasticity: Multiple mechanisms and functions - a lecture by Robert Malenka, M.D., Ph.D., Stanford University. Video podcast, runtime: 01:05:17.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||