Surface hopping
Surface hopping is a semi-classical technique that incorporates quantum mechanical effects into molecular dynamics simulations.[1][2][3][4] Traditional approaches propagate the dynamics only on one surface. However, quantum mechanics predicts that the dynamics happen on all the surfaces simultaneously. Surface hopping incorporates the effect of other surfaces by allowing for 'hops' between the surfaces, subject to certain criteria. This method is particularly useful when the Born-Oppenheimer approximation breaks down in certain regions, particularly conical intersections and avoided crossings.
Motivation
Molecular dynamics simulations numerically solve the classical equations of motion. These simulations, though, ignore the effects of zero-point energy, quantum interference, and quantum tunneling.[5] Solving the time-dependent Schrödinger equation numerically incorporates all these effects, but is computationally unfeasible when the system has many degrees of freedom. To tackle this issue, one approach is the mean field or Ehrenfest method, where the molecular dynamics is run on the average potential energy surface given by a linear combination of the adiabatic states. This was applied successfully for some applications, but has some important limitations. When the difference between the adiabatic states is large, then the dynamics must be primarily driven by only one surface, and not an average potential. In addition, this method also violates the principle of microscopic reversibility.[3]
Surface hopping accounts for these limitations by propagating an ensemble of trajectories, each one of them on a single adiabatic surface at any given time. The trajectories are allowed to 'hop' between various adiabatic states at certain times such that the quantum amplitudes for the adiabatic states follow the time dependent Schrödinger equation. The probability of these hops are dependent on the coupling between the states, and is generally significant only in the regions where the difference between adiabatic energies is small.
Theory behind the method
The formulation described here is in the adiabatic representation for simplicity.[5] It can easily be generalized to a different representation.
The coordinates of the system is divided into two categories: quantum (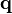 ) and classical (
) and classical (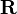 ). The Hamiltonian of the quantum degrees of freedom with mass
). The Hamiltonian of the quantum degrees of freedom with mass 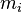 is defined as:
is defined as:
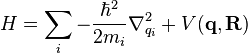 ,
,
where 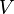 describes the potential for the whole system. The eigenvalues of
describes the potential for the whole system. The eigenvalues of 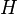 as a function of are called the adiabatic surfaces :
as a function of are called the adiabatic surfaces :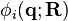 . Typically, corresponds to the electronic degree of freedom, light atoms such as hydrogen, or high frequency vibrations such as O-H stretch. The forces in the molecular dynamics simulations are derived only from one adiabatic surface, and are given by:
. Typically, corresponds to the electronic degree of freedom, light atoms such as hydrogen, or high frequency vibrations such as O-H stretch. The forces in the molecular dynamics simulations are derived only from one adiabatic surface, and are given by:
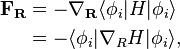
where i represents the chosen adiabatic surface. The last equation is derived using Hellmann-Feynman theorem. The brackets denote the integral is done only over the quantum degrees of freedom. Choosing only one adiabatic surface is an excellent approximation if the difference between the adiabatic surfaces is large for energetically accessible regions of . When this is not the case, the effect of the other states become important. This effect is incorporated in the surface hopping algorithm by considering the wavefunction of the quantum degrees of freedom at time t as an expansion in the adiabatic basis:
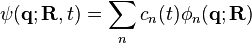 ,
,
where 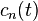 are the expansion coefficients. Substituting the above equation into the time dependent Schrödinger equation gives
are the expansion coefficients. Substituting the above equation into the time dependent Schrödinger equation gives
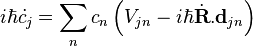 ,
,
where 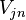 and the nonadiabatic coupling vector
and the nonadiabatic coupling vector 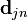 are given by
are given by
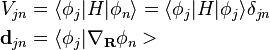
The adiabatic surface can switch at any given time t based on how the quantum probabilities 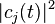 are changing with time. The rate of change of is given by:
are changing with time. The rate of change of is given by:
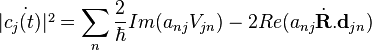 ,
,
where 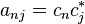 . For a small time interval dt, the fractional change in is given by
. For a small time interval dt, the fractional change in is given by
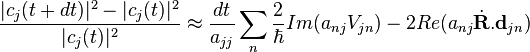 .
.
This gives the net change in flux of population from state 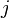 . Based on this, the probability of hopping from state j to n is proposed to be
. Based on this, the probability of hopping from state j to n is proposed to be
 .
.
This criteria is known as the "fewest switching" algorithm, as it minimizes the number of hops required to maintain the population in various adiabatic states.
Whenever a hop takes place, the velocity is adjusted to maintain conservation of energy. To compute the direction of the change in velocity, the nuclear forces in the transition is
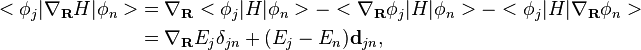
where 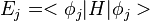 is the eigen value. For the last equality,
is the eigen value. For the last equality, 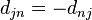 is used. This shows that the nuclear forces acting during the hop are in the direction of the nonadiabatic coupling vector . Hence is a reasonable choice for the direction along which velocity should be changed.
is used. This shows that the nuclear forces acting during the hop are in the direction of the nonadiabatic coupling vector . Hence is a reasonable choice for the direction along which velocity should be changed.
Frustrated Hops
If the velocity reduction required to conserve energy while making a hop is greater than the component of the velocity to be adjusted, then the hop is known as frustrated. In other words, a hop is frustrated if the system does not have enough energy to make the hop. Several approaches have been suggested to deal with these frustrated hops. The simplest of these is to ignore these hops. [2] Another suggestion is not to change the adiabatic state, but reverse the direction of the component of the velocity along the nonadiabatic coupling vector.[5] Yet another approach is to allow the hop to happen if an allowed hopping point is reachable within uncertainty time 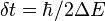 , where
, where 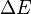 is the extra energy that the system needed to make the hop possible.[6]
is the extra energy that the system needed to make the hop possible.[6]
Decoherence Time
Surface hopping can develop nonphysical coherence between the quantum coefficients over large time. To eliminate this, the quantum coefficient is set to 1 for the current state (and zero for the rest of the states) after a predefined time has elapsed after the trajectory crosses the region where hopping has high probabilities. [5]
Outline of the algorithm
The state of the system at any time 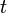 is given by the phase space of all the classical particles, the quantum amplitudes, and the adiabatic state. The simulation broadly consists of the following steps:
is given by the phase space of all the classical particles, the quantum amplitudes, and the adiabatic state. The simulation broadly consists of the following steps:
Step 1. Initialize the state of the system. The classical positions and velocities are chosen based on the ensemble required.
Step 2. Compute forces using Hellmann-Feynman theorem, and integrate the equations of motion by time step 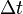 to obtain the classical phase space at time
to obtain the classical phase space at time 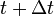 .
.
Step 3. Integrate the Schrödinger equation to evolve quantum amplitudes from time to in increments of 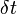 . This time step is typically much smaller than .
. This time step is typically much smaller than .
Step 4. Compute probability of hopping from current state to all other states. Generate a random number, and determine whether a switch should take place. If a switch does occur, change velocities to conserve energy. Go back to step 2, till trajectories have been evolved for the desired time.
Applications
The method has been applied successfully to understand dynamics of systems that include tunneling, conical intersections and electronic excitation.[7] [8]
Limitations
Most of the critique of the surface hopping method comes from the separation of classical and quantum degrees of freedom. This ignores the entanglement between these degrees of freedom. Moreover, this method is computationally feasible only for a limited number of quantum degrees of freedom. In addition, the trajectories must have enough energy to be able to reach the regions where probability of hopping is large.
See Also
- Molecular Dynamics
- Computational Chemistry
- Quantum Chemistry
- Path Integral Molecular Dynamics
References
- ↑ Herman, Michael F. (1984). "Nonadiabatic semiclassical scattering. I. Analysis of generalized surface hopping procedures". The Journal of Chemical Physics 81 (2): 754. Bibcode:1984JChPh..81..754H. doi:10.1063/1.447708.
- ↑ 2.0 2.1 Tully, John C. (1990). "Molecular dynamics with electronic transitions". The Journal of Chemical Physics 93 (2): 1061. Bibcode:1990JChPh..93.1061T. doi:10.1063/1.459170.
- ↑ 3.0 3.1 Quantum simulations of complex many-body systems: from theory to algorithms : winter school, 25 February - 1 March 2002, Rolduc Conference Centre, Kerkrade, the Netherlands ; lecture notes. Jülich: NIC-Secretariat. 2002. p. 377. ISBN 3-00-009057-6.
|first1=missing|last1=in Authors list (help) - ↑ Barbatti, Mario. "Nonadiabatic dynamics with trajectory surface hopping method". Wiley Interdisciplinary Reviews: Computational Molecular Science 1 (4): 620–633. doi:10.1002/wcms.64.
- ↑ 5.0 5.1 5.2 5.3 Hammes-Schiffer, Sharon; Tully, John C. (1994). "Proton transfer in solution: Molecular dynamics with quantum transitions". The Journal of Chemical Physics 101 (6): 4657. Bibcode:1994JChPh.101.4657H. doi:10.1063/1.467455.
- ↑ Jasper, Ahren W.; Stechmann, Samuel N.; Truhlar, Donald G. (2002). "Fewest-switches with time uncertainty: A modified trajectory surface-hopping algorithm with better accuracy for classically forbidden electronic transitions". The Journal of Chemical Physics 116 (13): 5424. Bibcode:2002JChPh.116.5424J. doi:10.1063/1.1453404.
- ↑ Jiang, Ruomu; Sibert, Edwin L. (2012). "Surface hopping simulation of vibrational predissociation of methanol dimer". The Journal of Chemical Physics 136 (22): 224104. Bibcode:2012JChPh.136v4104J. doi:10.1063/1.4724219.
- ↑ Müller, Uwe; Stock, Gerhard (22 October 1997). "Surface-hopping modeling of photoinduced relaxation dynamics on coupled potential-energy surfaces". The Journal of Chemical Physics 107 (16): 6230–6245. Bibcode:1997JChPh.107.6230M. doi:10.1063/1.474288.