Stetter reaction
The Stetter reaction is a reaction used in organic chemistry to form carbon-carbon bonds through a 1,4-addition reaction utilizing a nucleophilic catalyst.[1] While the related 1,2-addition reaction, the benzoin condensation, was known since the 1830s, the Stetter reaction was not reported until 1973 by Dr. Hermann Stetter.[2] The reaction provides synthetically useful 1,4-dicarbonyl compounds and related derivatives from aldehydes and Michael acceptors. Unlike 1,3-dicarbonyls, which are easily accessed through the Claisen condensation, or 1,5-dicarbonyls, which are commonly made using a Michael reaction, 1,4-dicarbonyls are challenging substrates to synthesize, yet are valuable starting materials for several organic transformations, including the Paal–Knorr synthesis of furans and pyrroles. Traditionally utilized catalysts for the Stetter reaction are thiazolium salts and cyanide anion, but more recent work toward the asymmetric Stetter reaction has found triazolium salts to be effective. The Stetter reaction is an example of umpolung chemistry, as the inherent polarity of the aldehyde is reversed by the addition of the catalyst to the aldehyde, rendering the carbon center nucleophilic rather than electrophilic.
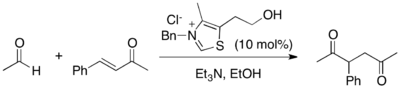
Mechanism
As the Stetter reaction is an example of umpolung chemistry, the aldehyde is converted from an electrophile to a nucleophile under the reaction conditions.[3] This is accomplished by activation from some catalyst - either cyanide (CN−) or thiazolium salt.[1] For the use of either catalyst, the mechanism is very similar; the only difference is that with thiazolium salts, the catalyst must be deprotonated first to form the active catalytic species. The active catalyst can be described as the combination of two contributing resonance forms - an ylide or a carbene, both of which portray the nucleophilic character at carbon. The thiazolium ylide or CN− can then add into the aldehyde substrate, forming a cyanohydrin in the case of CN− or the Breslow intermediate in the case of thiazolium salt. The Breslow intermediate was proposed by Ronald Breslow in 1958 and is a common intermediate for all thiamine-catalyzed reactions, whether in vitro or in vivo.[4]

Once the "nucleophilic aldehyde" synthon is formed, whether as a cyanohydrin or stabilized by a thiazolium ylide, the reaction can proceed down two pathways. The faster pathway is self-condensation with another molecule of aldehyde to give benzoin products. However, benzoin condensation is completely reversible, and therefore does not interfere with product formation in the Stetter reaction. In fact, benzoins can be used instead of aldehydes as substrates to achieve the same overall Stetter transformation, because benzoins can be restored to their aldehyde precursors under the reaction conditions.[1] The desired pathway toward the Stetter product is the 1,4-addition of the nucleophilic aldehyde to a Michael-type acceptor. After 1,4-addition, the reaction is irreversible and ultimately, the 1,4-dicarbonyl is formed when the catalyst is kicked out to regenerate CN− or the thiazolium ylide.

Scope
The Stetter reaction produces classically difficult to access 1,4-dicarbonyl compounds and related derivatives. The traditional Stetter reaction is quite versatile, working on a wide variety of substrates.[1] Aromatic aldehydes, heteroaromatic aldehydes, and benzoins can all be used as acyl anion precursors with thiazolium salt and cyanide catalysts. However, aliphatic aldehydes can only be utilized if a thiazolium salt is used as a catalyst, as they undergo aldol condensation side reaction when a cyanide catalyst is used. In addition, α,β-unsaturated esters, ketones, nitriles, nitros, and aldehydes are all appropriate Michael acceptors with either catalyst. However, the general scope of asymmetric Stetter reactions is more limited. Intramolecular asymmetric Stetter reactions enjoy a range of acceptable Michael acceptors and acyl anion precursors in essentially any combination.[5] Intramolecular asymmetric Stetter reactions can utilize aromatic, heteroaromatic and aliphatic aldehydes with a tethered α,β-unsaturated ester, ketone, thioester, malonate, nitrile or Weinreb amide. It has been shown that α,β-unsaturated nitros and aldehydes are not suitable Michael acceptors and have markedly decreased enantiomeric excess in such reactions.[5] Another limitation encountered with intramolecular asymmetric Stetter reactions is that only substrates that result in the formation of a six-membered ring show synthetically useful enantiomeric excess; substrates which form five and seven-membered rings either do not react or show low stereoinduction.[5] On the other hand, intermolecular asymmetric reactions are quite confined to specifically matched combinations of acyl anion precursor and Michael acceptor, such as an aliphatic aldehyde with a nitroalkene.[6] In addition, these substrates tend to be rather activated, as the intermolecular asymmetric Stetter reaction is still in the early stages of development.

Variations
Several variations of the Stetter reaction have been developed since its discovery in 1973. In 2001, Murry et al reported a Stetter reaction of aromatic aldehydes onto acylimine derivatives to give α-amido ketone products.[7] The acylimine acceptors were generated in situ from α-tosylamide substrates, which underwent elimination in the presence of base. Good to excellent yields (75-90%) were observed. Mechanistic investigations showed that the corresponding benzoins were not adequate substrates, contrary to traditional Stetter reactions.[1] From this, the authors conclude the Stetter reaction of acylimines is under kinetic control, rather than thermodynamic control.
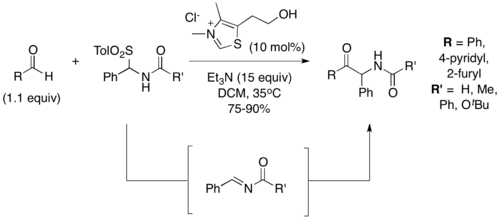
Another variation of the Stetter reaction involves the use of 1,2-dicarbonyls as precursors to the acyl anion intermediate. In 2005, Scheidt and coworkers reported the use of sodium pyruvate, which loses CO2 to form the Breslow intermediate.[8] Similarly, in 2011 Bortolini and coworkers demonstrated the use of α-diketones to generate an acyl anion.[9] Under the conditions they developed, 2,3-butadienone is cleaved after addition to the thiazolium catalyst to release ethyl acetate and generate the Breslow intermediate necessary for the Stetter reaction to proceed.
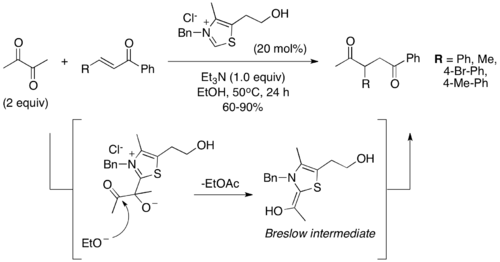
In addition, they showed the atom economy and utility of using a cyclic α-diketone to generate the Stetter product with a tethered ethyl ester. The reaction precedes through the same mechanism as the acyclic version, but the ester generated by attack of ethanol remains tethered to the product. However, the conditions only allow for the generation of ethyl esters, due to the necessity of ethanol as solvent. Substitution of ethanol with tert-butanol resulted in no product. The authors speculate this is due to the difference in acidity between the two alcoholic solvents.

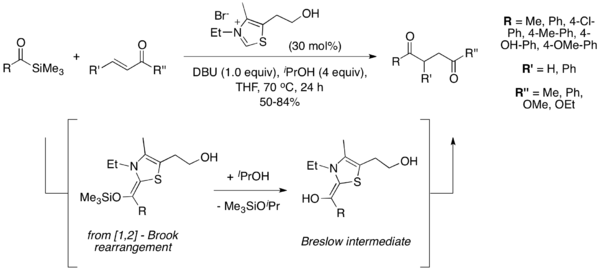
In 2004, Scheidt and coworkers introduced acyl silanes as competent substrates in the Stetter reaction, a variation they termed the "sila-Stetter reaction."[10] Under their reaction conditions, the thiazolium catalyst induces a [1,2] Brook rearrangement, which is followed by desilylation by an isopropanol additive to give the common Breslow intermediate of the traditional Stetter reaction. The desilylation step was found to be necessary, and the reaction does not proceed without an alcoholic additive. Acyl silanes are less electrophilic than the corresponding aldehydes, preventing typical benzoin-type byproducts often observed in the Stetter reaction.[11]
Asymmetric Stetter Reaction
The first asymmetric variant of the Stetter reaction was reported in 1996 by Enders et al, employing a chiral triazolium catalyst 1.[12] Subsequently, several other catalysts were reported for asymmetric Stetter reactions, including 2,[13] 3,[14] and 4.[15]

The success of the Rovis group's catalyst 2 led them to further explore this family of catalysts and expand their use for asymmetric Stetter reactions. In 2004, they reported the enantioselective formation of quaternary centers from aromatic aldehydes in an intramolecular Stetter reaction with a slightly modified catalyst.[16] Further work extended the scope of this reaction to include aliphatic aldehydes as well.[17] Subsequently, it was shown that the olefin geometry of the Michael acceptor dictates diastereoselectivity in these reactions, whereby the catalyst dictates the enantioselectivity of the initial carbon bond formation and allylic strain minimization dictates the diastereoselective intramolecular protonation.[18]
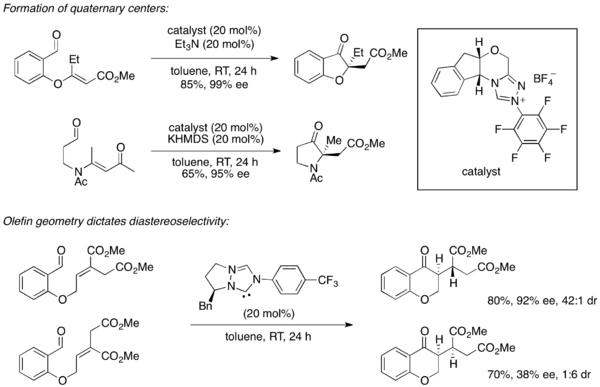
The inherent difficulties of controlling enantioselectivity in intermolecular reactions made the development of an intermolecular asymmetric Stetter reaction a challenge. While limited enantiomeric excess had been reported by Enders in the early 1990s for the reaction of n-butanal with chalcone,[19] conditions for a synthetically useful asymmetric intermolecular Stetter reaction were not reported until 2008 when both the groups of Enders and Rovis published such reactions. The Enders group utilized a triazolium-based catalyst to effect the coupling of aromatic aldehydes with chalcone derivatives with moderate yields.[20] The concurrent publication from the Rovis group also employed a triazolium-based catalyst and reported the Stetter reaction between glyoxamides and alkylidenemalonates in good to excellent yields.[21]

Rovis and coworkers subsequently went on to explore the asymmetric intermolecular Stetter reaction of heterocyclic aldehydes and nitroalkenes.[22] During optimization of this reaction, it was found that a catalyst with a fluorinated backbone greatly enhanced enantioselectivity in the reaction. It was proposed that the fluorinated backbone helps to lock the conformation of the catalyst in a way the increases enantioselectivity. Further computational studies on this system verified that the stereoelectronic attraction between the developing partial negative charge on the nitroalkene in the transition state and the partial positive charge of the C-F dipole is responsible for the increase in enantiomeric excess observed with the use of the catalyst with backbone fluorination.[23] While this is a marked advance in the area of intermolecular asymmetric Stetter reactions, the substrate scope is limited and the catalyst is optimized for the specific substrates being utilized.

Another contribution to the development of asymmetric intermolecular Stetter reactions came from Glorius and coworkers in 2011.[6] They demonstrated the synthesis of α-amino acids enantioselectively by utilizing N-acylamido acrylate as the conjugate acceptor. Significantly, the reaction can be run on a 5 mmol scale without loss of yield or enantioselectivity.

Applications
The Stetter reaction is an effective tool in organic synthesis. The products of the Stetter reaction, 1,4-dicarbonyls, are valuable moieties for the synthesis of complex molecules. For example, Trost and coworkers employed a Stetter reaction as one step in their synthesis of rac-hirsutic acid C.[24] The intramolecular coupling of an aliphatic aldehyde with a tethered α,β-unsaturated ester led to the desired tricyclic 1,4-dicarbonyl in 67% yield. This intermediate was converted into rac-hirsutic acid C in seven more steps.

The Stetter reaction is commonly used in sequence with the Paal-Knorr synthesis of furans and pyrroles, which a 1,4-dicarbonyl undergoes condensation with itself or in the presence of an amine under high temperature, acidic conditions. In 2001, Tius and coworkers reported the asymmetric total synthesis of roseophilin utilizing an intermolecular Stetter reaction to couple an aliphatic aldehyde with a cyclic enone.[25] After ring-closing metathesis and alkene reduction, the 1,4-dicarbonyl product was converted to a pyrrole via the Paal-Knorr synthesis and further elaborated to the natural product.

In 2004, a one-pot coupling-isomerization-Stetter-Paal Knorr sequence was reported.[26] This procedure first utilizes palladium cross-coupling chemistry to couple aryl halides with propargylic alcohols to give α,β-unsaturated ketones, which can then undergo a Stetter reaction with an aldehyde. Once the 1,4-dicarbonyl compound is formed, heating in the presence of acid will give the furan, while heating in the presence of ammonium chloride and acid will give the pyrrole. The entire sequence is performed in one-pot with no work-up or purification between steps.
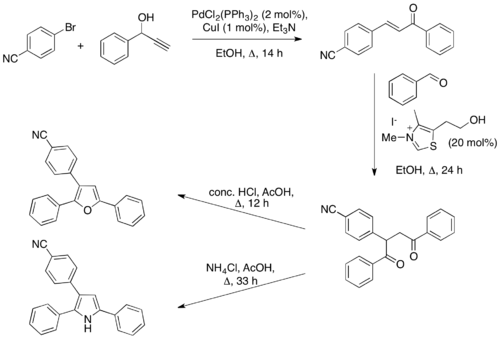
Ma and coworkers developed an alternative method for accessing furans utilizing the Stetter reaction.[27] In their report, 3-aminofurans are synthesized under Stetter conditions for coupling aromatic aldehydes with dimethyl acetylenedicarboxylate (DMAD), whereby the thiazolium ylide is hydrolyzed by the aromatization of the furan product. As the thiazolium is destroyed under these conditions, it is not catalytic and must be used in stoichiometric quantities.

They further elaborated on this work by developing a method in which 2-aminofurans are synthesized by cyclization onto a nitrile.[28] In this method, the thiazolium ylide is employed catalytically and the free amine product is generated.

Related
- Benzoin condensation
- Thiamine
- Baylis–Hillman reaction
- Paal-Knorr synthesis
- Umpolung
References
- ↑ 1.0 1.1 1.2 1.3 1.4 Stetter, H. Angew. Chem. Int. Ed. 1976, 15, 639.
- ↑ Stetter, H. and Schreckenberg, M. Angew. Chem. Int. Ed. Engl. 1973, 12, 81.
- ↑ Albright, J. D. Tetrahedron 1983, 39, 3207.
- ↑ Breslow, R. J. Am. Chem. Soc. 1958, 80, 3719.
- ↑ 5.0 5.1 5.2 de Alaniz, J. R.; Kerr, M. S.; Moore, J. L.; Rovis, T. J. Org. Chem. 2008, 73, 2033.
- ↑ 6.0 6.1 Jousseaume, T.; Wurz, N. E.; Glorius, F. Angew. Chem. Int. Ed. 2011, 50, 1410.
- ↑ Murry, J. A.; Frantz, D. E.; Soheili, A.; Tillyer, R.; Grabowski, E. J. J.; Reider, P. J. J. Am. Chem. Soc. 2001, 123, 9696.
- ↑ Myers, M. C.; Bharadwaj, A. R.; Milgram, B. C.; Scheidt, K. A. J. Am. Chem. Soc. 2005, 127, 14675.
- ↑ Bortolini, O.; Fantin, G.; Fogagnolo, M.; Giovannini, P. P.; Massi, A.; Pacifico, S. Org. Biomol. Chem. 2011, 9, 8437.
- ↑ Mattson, A. E.; Bharadwaj, A. R.; Scheidt, K. A. J. Am. Chem. Soc. 2004, 126, 2314.
- ↑ Mattson, A. E.; Bharadwaj, A. R.; Zuhl, A. M.; Scheidt, K. A. "Thiazolium-Catalyzed Additions of Acylsilanes: A General Strategy for Acyl Anion Addition Reactions." J. Org. Chem. 2006, 71, 5715. doi:10.1021/jo060699c
- ↑ Enders, D.; Breuer K.; Runsink, J.; Teles, J. H. Helv. Chim. Acta 1996, 79, 1899.
- ↑ Kerr, M. S.; de Alaniz, J. R.; Rovis, T. J. Am. Chem. Soc. 2002, 124, 10298.
- ↑ Pesch, J.; Harms, K.; Bach, T. Eur. J. Org. Chem. 2004, 2025.
- ↑ Mennen, S. M.; Blank, J. T.; Tran-Dubé, M. B.; Imbriglio, J. E.; Miller, S. J. Chem. Commun. 2005, 195.
- ↑ Kerr, M. S.; Rovis, T. J. Am. Chem. Soc. 2004, 126, 8876.
- ↑ Moore, J. L.; Kerr, M. S.; Rovis, T. Tetrahedron 2006, 62, 11477.
- ↑ de Alaniz, J. R.; Rovis, T. J. Am. Chem. Soc. 2005, 127, 6284.
- ↑ Enders, D. Enzymemimetic C-C and C-N Bond Formations. In Stereoselective Synthesis; Ottow, E., Schoellkopf, K., Schulz, B.-G., eds.; Springer-Verlag: Berlin-Heidelberg, 1994; pp 63-90.
- ↑ Enders, D.; Han, J.; Henseler, A. Chem. Commun. 2008, 3989.
- ↑ Liu, Q.; Perreault, S.; Rovis, T. J. Am. Chem. Soc. 2008, 130, 14066.
- ↑ DiRocco, D. A.; Oberg, K. M.; Dalton, D. M.; Rovis, T. J. Am. Chem. Soc. 2009, 131, 10872.
- ↑ Um, J. M.; DiRocco, D. A.; Noey, E. L.; Rovis, T.; Houk, K. N. J. Am. Chem. Soc. 2011, 133, 11249.
- ↑ Trost, B.M.; Shuey, C. D.; DiNinno, F., Jr.; McElvain, S. S. J. Am. Chem. Soc. 1979, 101, 1284.
- ↑ Harrington, P. E.; Tius, M. A. J. Am. Chem. Soc. 2001, 123, 8509.
- ↑ Braun, R. U.; Müller, T. J. J. Synthesis 2004, 14, 2391.
- ↑ Ma, C.; Yang, Y. Org Lett. 2005, 7,1343.
- ↑ Liu, P.; Lei, M.; Ma, L.; Hu, L. Synlett 2011, 8, 1133.