Stereochemistry of ketonization of enols and enolates
In the stereochemistry of ketonization of enols and enolates, theory is provided explaining the diastereoselectivity[1] observed in the conversion of certain enols and enolates into the corresponding ketone.
Introduction
Ketones and their corresponding enols are isomers, termed tautomers. These are easily interconvertible. But simple enols are not generally stable and of considerably higher energy than the corresponding ketones. Nevertheless, a very large number of organic reactions proceed via enolic intermediates. Thus the behavior of enols is critical to an understanding of myriad organic reactions.
Many of these enols, formed in an organic reaction, a priori, can lead onward and afford two diastereomers on ketonization. If one knows the stereochemistry of ketonization of these enolic intermediates, then one can predict the stereochemistry of myriad organic reactions.
It was proposed in 1955[2] that the kinetic protonation of enolic species proceeds with an early transition state with the alpha carbon being close to sp2 hybridized. The proton donor selectively approaches the less hindered face of the enolate, thus leading to the less stable of two diastereomers. Reactions controlled in this fashion include:
- (a) decarboxylation of malonic acids
- (b) decarboxylation of beta-keto acids
- (c) the Michael addition of nucleophiles to unsaturated carbonyl compounds
- (d) the Birch reduction of enones
- (e) the deprotonation – protonation of carbonyl compounds
- (f) the dehalogenation of alpha-haloketones
- (f) the Norrish Type II Reaction
- and many more.[2][3][4][5][6][7][8][9]
Kinetic or thermodynamic control
Protonation from the less hindered face of an enol leads to the less stable of two, a priori, diastereomers. In this example[6] there are two different reactions which afford the enol as a transient intermediate. One is the treatment of an α-bromoketone with dilute HI in acetone. The second is the reaction of an enol acetate with methyllithium. The first of the two reactions is an example of microscopic reversibility. This is the reverse of bromination of a ketone, a reaction well-known to proceed via the enol as an intermediate. Interestingly this is an example with extreme stereoselectivity due to the severe steric hindrance of an ethano-bridge.
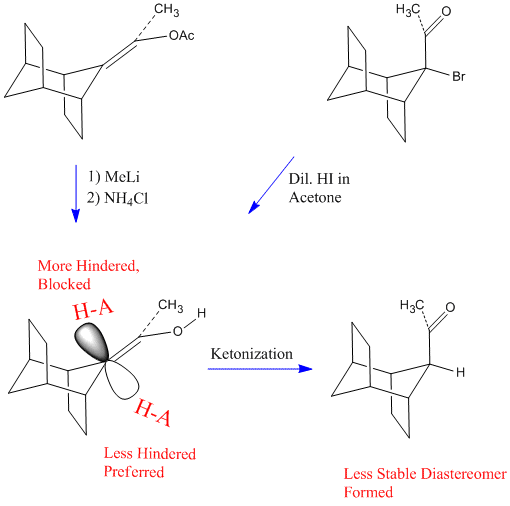
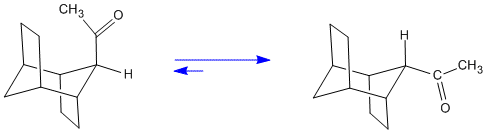
With an acid catalyst as well as with a base catalyst such as sodium ethoxide a thermodynamic equilibrium is achieved. The diastereomer formed now has the acetyl group equatorial.
Figure 2. Equilibration of the Diastereomers via the Common Enol.
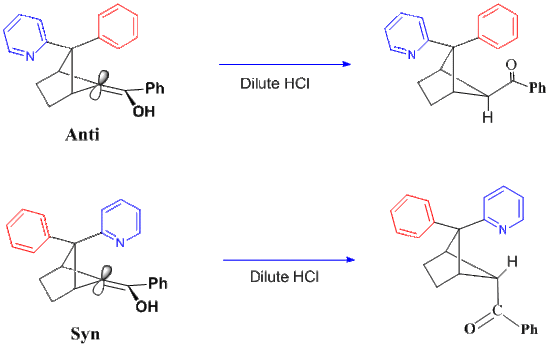
Unusual case of a phenyl pyridyl enol
Figure 3 show the ketonization results for the two Phenyl-Pyridyl diastereomers. In the exo-pyridyl isomer on the left, the usual steric hindrance control blocks protonation from above. That is, the phenyl group is positioned directly above the enolic alpha carbon and protonation must occur from below. In contrast, in the case of the endo pyridyl isomer on the right, the basic pyridyl moiety proves capable of picking up the proton first and then delivering it to the alpha-carbon from this upper, hindered side. The result from intramolecular proton delivery is the reverse of the common stereochemistry.[9]
Figure 3. Two Phenyl-Pyridyl Enol Diastereomers.
A Typical example; an enol generated from an alpha-bromoketone
Figure 4. The Example of Ketonization of the Enol of 4-Phenyl-1-Benzoylcyclohexane.

In this example the enol intermediate is generated from either the cis- or the trans-stereoisomer of 1-bromo-1-benzoyl-4-phenylcyclohexane using zinc as the reagent. The endo proton approach is blocked by two axial hydrogens. This example is somewhat more typical than those shown earlier since the stereoselectivity[1] is only in the range of 60 to 70 percent favoring formation of the (less stable) cis product.[10]
References
- ↑ 1.0 1.1 (a)"Overlap Control of Carbanionoid Reactions. I. Stereoselectivity in Alkaline Epoxidation," Zimmerman, H. E.; Singer, L.; Thyagarajan, B. S. J. Am. Chem. Soc., 1959, 81, 108-116.(b)Eliel, E., "Stereochemistry of Carbon Compound", McGraw-Hill, 1962 pp 434-436.
- ↑ 2.0 2.1 "The Stereochemistry of the Ketonization Reaction of Enols," Zimmerman, H. E. J. Org. Chem., 1955, 20, 549-557.
- ↑ (a) "The Stereochemistry of the Ketonization Reaction of Enols. III," Zimmerman, H. E.; Giallombardo, H. J. J. Am. Chem. Soc., 1956, 78, 6259-6263.
- ↑ (b) "The Stereochemistry of Ketonization. IV," Zimmerman, H. E. J. Am. Chem. Soc., 1957, 79, 6554-6558.,
- ↑ (c) "The Stereochemistry of Ketonization. X. Enols from alpha-Haloacids," Zimmerman, H. E.; Cutshall, T. W. J. Am. Chem. Soc., 1959, 81, 4305-4308.
- ↑ 6.0 6.1 6.2 (d) "Enhanced Endo-Exo Selectivity in the Stereochemistry of Ketonization of Enols", Zimmerman, H. E.; Linder, L. W. J. Org. Chem., 1985,c48, 1637-1646.
- ↑ (e) "Kinetic Protonation of Enols, Enolates and Analogs; The Stereochemistry of Ketonization," Zimmerman, H. E. Accounts of Chem. Res., 1987, 20, 263-268.
- ↑ (f) “The Stereochemistry of Allenic Enol Tautomerism; Independent Generation and Reactivity” Zimmerman, H. E.; Pushechnikov, A., Eur J. Org. Chem., 2006, 15 3491-3497.
- ↑ 9.0 9.1 (g) “Inter and Intramolecular Stereoselective Protonation of Enols”, Zimmerman, H. E.; Wang, P., J. Org. Chem. 2002, 69, 9216-9226.
- ↑ "The Stereochemistry of Protonation. XI," Zimmerman, H. E.; Mariano, P. S., J. Am. Chem. Soc., 1968, 90, 6091-6096.