Specific ion interaction theory
Specific ion Interaction Theory (SIT theory) is a theory used to estimate single-ion activity coefficients in electrolyte solutions at relatively high concentrations.[1][2] It does so by taking into consideration interaction coefficients between the various ions present in solution. Interaction coefficients are determined from equilibrium constant values obtained with solutions at various ionic strengths. The determination of SIT interaction coefficients also yields the value of the equilibrium constant at infinite dilution.
Background
The need for this theory arises from the need to derive activity coefficients of solutes when their concentrations are too high to be predicted accurately by Debye-Hückel theory. These activity coefficients are needed because an equilibrium constant is defined in thermodynamics as a quotient of activities but is usually measured using concentrations. The protonation of a monobasic acid will be used to simplify the exposition. The equilibrium for protonation of the conjugate base, A− of the acid, may be written as
- A− + H+
 AH
AH
for which
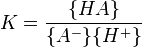
where {HA} signifies an activity of the chemical species HA etc.. The role of water in the equilibrium has been ignored as in all but the most concentrated solutions the activity of water is a constant. Note that K is defined here as an association constant, the reciprocal of an acid dissociation constant.
Each activity term can be expressed as the product of a concentration and an activity coefficient. For example,
- {HA} = [HA] × γHA
where the square brackets signify a concentration and γ is an activity coefficient. Thus the equlilbrim constant can be expressed as a product of a concentration quotient and an activity coefficient quotient.
![K=\frac{[HA]}{[H^+][A^-]}\times \frac{\gamma_{HA}}{\gamma_{H^+}\gamma_{A^-}}](../I/m/b045be8a57e371d8d754820ba31e670b.png)
Taking logarithms.
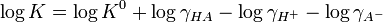
K0 is the hypothetical value that the equilibrium constant would have if the solution of the acid were so dilute that the activity coefficients were all equal to one.
It is common practise to determine equilibrium constants in solutions containing an electrolyte at high ionic strength such that the activity coefficients are effectively constant. However, when the ionic strength is changed the measured equilibrium constant will also change, so there is a need to estimate individual (single ion) activity coefficients. Debye-Huckel theory provides a means to do this, but it is accurate only at very low concentrations. Hence the need for an extension to Debye-Hückel theory. Two main approaches have been used. SIT theory, discussed here and Pitzer equations.[3][4]
Development
SIT theory was first proposed by Brønsted[5] and was further developed by Guggenheim.[1] Scatchard.[6] extended the theory to allow the interaction coefficients to vary with ionic strength. The theory was mainly of theoretical interest until 1945 because of the difficulty of determining equilibrium constants before the glass electrode was invented. Subsequently Ciavatta[2] developed the theory further.
The activity coefficient of the jth ion in solution is written as γj when concentrations are on the molal concentration scale and as yj when concentrations are on the molar concentration scale. (The molality scale is preferred in thermodynamics because molal concentrations are independent of temperature). The basic idea of SIT theory is that the activity coefficient can be expressed as
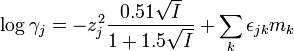 (molalities)
(molalities)
or
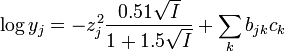 (molar concentrations)
(molar concentrations)
where z is the electrical charge on the ion, I is the ionic strength, ε and b are interaction coefficients and m and c are concentrations. The summation extends over the other ions present in solution, which includes the ions produced by the background electrolyte. The first term in these expressions comes from Debye-Hückel theory. The second term shows how the contributions from "interaction" are dependent on concentration. Thus, the interaction coefficients are used as corrections to Debye-Hückel theory when concentrations are higher than the region of validity of that theory.
The activity coefficient of a neutral species can be assumed to depend linearly on ionic strength, as in
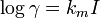
where km is a Setschenow coefficient.[7]
In the example of a monobasic acid HA, assuming that the background electrolyte is the salt NaNO3, the interaction coefficients will be for interaction between H+ and NO3−, and between A− and Na+.
Determination and application
Firstly, equilibrium constants are determined at a number of different ionic strengths, at a chosen temperature and particular background electrolyte. The interaction coefficients are then determined by fitting to the observed equilibrium constant values. The procedure also provides the value of K at infinite dilution. It is not limited to monobasic acids.[8] and can also be applied to metal complexes.[9] The SIT and Pitzer approaches have been compared recently.[10] The Bromley equation[11] has also been compared to both SIT and Pitzer equations.[12]
References
- ↑ 1.0 1.1 Guggenheim, E.A.; Turgeon, J.C. (1955). "Specific interaction of ions". Trans. Faraday Soc. 51: 747–761. doi:10.1039/TF9555100747.
- ↑ 2.0 2.1 Ciavatta, L. (1980). "The specific interaction theory in the evaluating ionic equilibria". Ann. Chim. (Rome) 70: 551–562.
- ↑ Pitzer, K.S. (1973). "Thermodynamics of electrolytes, I. Theoretical basis and general equations". J. Phys. Chem. 77: 268–277. doi:10.1021/j100621a026.
- ↑ Pitzer, K.S. (1991). Activity coefficients in electrolyte solutions. Boca Raton, Fla: CRC Press. ISBN 0-8493-5415-3.
- ↑ Brønsted, J.N. (1922). "Studies on solubility IV. The principle of the specific interaction of ions". J. Am. Chem. Soc. 44: 877–898. doi:10.1021/ja01426a001.
- ↑ Scatchard, G. (1936). "Concentated solutions of strong electrolytes". Chem. Rev. 19: 309–327. doi:10.1021/cr60064a008.
- ↑ Setchenow, I.M. (1892). Ann. Chim. Phys. 25: 226. Missing or empty
|title=(help) - ↑ Crea, F.; De Stefano, C.; Foti, C.; Sammartano, S. (2007). "Sit parameters for the dependence of (poly)carboxylate activity coefficients on ionic strength ...". J. Chem. Eng. Data 52: 2195–2203. doi:10.1021/je700223r.
- ↑ Ciavatta, L. (1990). "The specific interaction theory in equilibrium analysis. Some empirical rules for estimate interaction coefficients of metal ion complexes". Ann. Chim. (Rome) 80: 255–263.
- ↑ Elizalde, M. P.; Aparicio, J. L. (1995). "Current theories in the calculation of activity coefficients—II. Specific interaction theories applied to some equilibria studies in solution chemistry". Talanta 42 (3): 395–400. doi:10.1016/0039-9140(95)01422-8. PMID 18966243.
- ↑ Bromley, L.A. (1973). "Thermodynamic properties of strong electrolytes in aqueous solutions". AIChEJ 19 (2): 313–320. doi:10.1002/aic.690190216.
- ↑ Foti, C.; Gianguzza, A.; Sammartano, S. (1997). "Comparison of equations for fitting protonation constants of carboxylic acids in aqueous tetramethylammonium chloride at various ionic strengths". Journal of Solution Chemistry 26 (6): 631–648. doi:10.1007/BF02767633.
External links
- SIT program A PC program to correct stability constants for changes in ionic strength using SIT theory and to estimate SIT parameters with full statistics. Contains an editable database of published SIT parameters. It also provides routines to inter-convert MolaRities (c) and MolaLities (m), and lg K(c) and lg K(m).