Similarity matrix
A similarity matrix is a matrix of scores that represent the similarity between a number of data points. Each element of the similarity matrix contains a measure of similarity between two of the data points. Similarity matrices are strongly related to their counterparts, distance matrices and substitution matrices.
Uses
Similarity matrices have a wide range of uses:
- To find clusters of data points.
- To align sequences of DNA.
Use in clustering
In spectral clustering, a similarity, or affinity, matrix is used to transform data to overcome difficulties related to lack of convexity in the shape of the data distribution.[1] The value of point 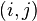 in the matrix can be simply the (negative of the) euclidean distance between
in the matrix can be simply the (negative of the) euclidean distance between 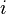 and
and 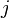 , or it can be a more complex measure of distance such as the Gaussian
, or it can be a more complex measure of distance such as the Gaussian 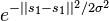 .[1] Further modifying this result with network analysis techniques is also common.[2]
.[1] Further modifying this result with network analysis techniques is also common.[2]
Use in sequence alignment
Similarity matrices are used in sequence alignment. Higher scores are given to more-similar characters, and lower or negative scores for dissimilar characters.
Nucleotide similarity matrices are used to align nucleic acid sequences. Because there are only four nucleotides commonly found in DNA (Adenine (A), Cytosine (C), Guanine (G) and Thymine (T)), nucleotide similarity matrices are much simpler than protein similarity matrices. For example, a simple matrix will assign identical bases a score of +1 and non-identical bases a score of −1. A more complicated matrix would give a higher score to transitions (changes from a pyrimidine such as C or T to another pyrimidine, or from a purine such as A or G to another purine) than to transversions (from a pyrimidine to a purine or vice versa). The match/mismatch ratio of the matrix sets the target evolutionary distance.[3][4] The +1/−3 DNA matrix used by BLASTN is best suited for finding matches between sequences that are 99% identical; a +1/−1 (or +4/−4) matrix is much more suited to sequences with about 70% similarity. Matrices for lower similarity sequences require longer sequence alignments.
Amino acid similarity matrices are more complicated, because there are 20 amino acids coded for by the genetic code, and so a larger number of possible substitutions. Therefore, the similarity matrix for amino acids contains 400 entries (although it is usually symmetric). The first approach scored all amino acid changes equally. A later refinement was to determine amino acid similarities based on how many base changes were required to change a codon to code for that amino acid. This model is better, but it doesn't take into account the selective pressure of amino acid changes. Better models took into account the chemical properties of amino acids.
One approach has been to empirically generate the similarity matrices. The Dayhoff method used phylogenetic trees and sequences taken from species on the tree. This approach has given rise to the PAM series of matrices. PAM matrices are labelled based on how many nucleotide changes have occurred, per 100 amino acids. While the PAM matrices benefit from having a well understood evolutionary model, they are most useful at short evolutionary distances (PAM10 - PAM120). At long evolutionary distances, for example PAM250 or 20% identity, it has been shown that the BLOSUM matrices are much more effective.
The BLOSUM series were generated by comparing a number of divergent sequences. The BLOSUM series are labeled based on how much entropy remains unmutated between all sequences, so a lower BLOSUM number corresponds to a higher PAM number.
See also
- Recurrence plot, a visualisation tool of recurrences in dynamical (and other) systems
- Self-similarity matrix
- Similarity testing
Notes and references
- ↑ 1.0 1.1 Ng, A.Y.; Jordan, M.I.; Weiss, Y. (2001), "On Spectral Clustering: Analysis and an Algorithm" (PDF), Advances in Neural Information Processing Systems (MIT Press) 14: 849–856
- ↑ Li, Xin-Ye; Guo, Li-Jie (2012), "Constructing affinity matrix in spectral clustering based on neighbor propagation", Neurocomputing (MIT Press) 97: 125–130, doi:10.1016/j.neucom.2012.06.023
- ↑ States, D; Gish, W; Altschul, S (1991). "Improved sensitivity of nucleic acid database searches using application-specific scoring matrices". Methods: a companion to methods in enzymology 3 (1): 66. doi:10.1016/S1046-2023(05)80165-3.
- ↑ Sean R. Eddy (2004). "Where did the BLOSUM62 alignment score matrix come from?" (PDF). Nature Biotechnology 22 (8): 1035–6. doi:10.1038/nbt0804-1035. PMID 15286655.