STO-nG basis sets
STO-nG basis sets are minimal basis sets, where 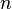 primitive Gaussian orbitals are fitted to a single Slater-type orbital (STO). originally took the values 2 - 6. They were first proposed by John Pople. A minimum basis set is where only sufficient orbitals are used to contain all the electrons in the neutral atom. Thus for the hydrogen atom, only a single 1s orbital is needed, while for a carbon atoms, 1s, 2s and three 2p orbitals are needed. The core and valence orbitals are represented by the same number of primitive Gaussian functions
primitive Gaussian orbitals are fitted to a single Slater-type orbital (STO). originally took the values 2 - 6. They were first proposed by John Pople. A minimum basis set is where only sufficient orbitals are used to contain all the electrons in the neutral atom. Thus for the hydrogen atom, only a single 1s orbital is needed, while for a carbon atoms, 1s, 2s and three 2p orbitals are needed. The core and valence orbitals are represented by the same number of primitive Gaussian functions 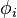 . For example, an STO-3G basis set for the 1s, 2s and 2p orbital of the carbon atom are all linear combination of 3 primitive Gaussian functions. For example, a STO-3G s orbital is given by:
. For example, an STO-3G basis set for the 1s, 2s and 2p orbital of the carbon atom are all linear combination of 3 primitive Gaussian functions. For example, a STO-3G s orbital is given by:
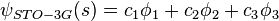
where
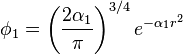
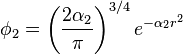
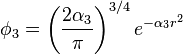
The values of c1, c2, c3, α1, α2 and α3 have to be determined. For the STO-nG basis sets, they are obtained by making a least squares fit of the three Gaussian orbitals to the single Slater-type orbitals. This differs from the more common procedure where the criteria often used is to choose the coefficients (c's) and exponents (α's) to give the lowest energy with some appropriate method for some appropriate molecule. A special feature of this basis set is that common exponents are used for orbitals in the same shell (e.g. 2s and 2p) as this allows more efficient computation.[1]
The fit between the Gaussian orbitals and the Slater orbital is good for all values of r, except for very small values near to the nucleus. The Slater orbital has a cusp at the nucleus, while Gaussian orbitals are flat at the nucleus.[2][3]
Use of STO-nG basis sets
The most widely used basis set of this group is STO-3G, which is used for large systems and for preliminary geometry determinations. This basis set is available for all atoms from Hydrogen up to Xenon.[4]
STO-2G basis set
The STO-2G basis set is a linear combination of 2 primitive Gaussian functions. The original coefficients and exponents for first-row and second-row atoms are given as follows.[1]
| STO-2G | α1 | c1 | α2 | c2 |
| 1s | 0.151623 | 0.678914 | 0.851819 | 0.430129 |
| 2s | 0.0974545 | 0.963782 | 0.384244 | 0.0494718 |
| 2p | 0.0974545 | 0.61282 | 0.384244 | 0.511541 |
Accuracy
The exact energy of the 1s electron of H atom is -0.5 hartree, given by a single Slater-type orbital with exponent 1.0. The following table illustrates the increase in accuracy as the number of primitive Gaussian functions increases from 3 to 6 in the basis set.[1]
| Basis set | Energy [hartree] |
| STO-3G | -0.49491 |
| STO-4G | -0.49848 |
| STO-5G | -0.49951 |
| STO-6G | -0.49983 |
See also
- List of quantum chemistry and solid state physics software
References
- ↑ 1.0 1.1 1.2 Hehre, W. J.; R. F. Stewart; J. A. Pople (1969). "Self-Consistent Molecular-Orbital Methods. I. Use of Gaussian Expansions of Slater-Type Atomic Orbitals". Journal of Chemical Physics 51 (6): 2657–2664. Bibcode:1969JChPh..51.2657H. doi:10.1063/1.1672392.
- ↑ Chemical Modeling From Atoms to Liquids, Alan Hinchliffe, John Wiley & Sons, Ltd., 1999. pg 294.
- ↑ Molecular Modelling, Andrew R. Leach, Longman, 1996. pg 68 - 73.
- ↑ Computational Chemistry, David Young, Wiley-Interscience, 2001. pg 86.