Ring-closing metathesis
Ring-closing metathesis, or RCM, is a widely used variation of olefin metathesis in organic chemistry for the synthesis of various unsaturated rings via the intramolecular metathesis of two terminal alkenes, which forms the cycloalkene as the E- or Z- isomers and volatile ethylene.[1][2]

The most commonly synthesized ring sizes are between 5-7 atoms;[3] however, reported syntheses include 45- up to 90- membered macroheterocycles.[4][5][6] These reactions are metal-catalyzed and proceed through a metallacyclobutane intermediate.[7] It was first published by Dider Villemin in 1980 describing the synthesis of an Exaltolide precursor,[8] and later become popularized by Robert H. Grubbs and Richard R. Schrock, who shared the Nobel Prize in Chemistry, along with Yves Chauvin, in 2005 for their combined work in olefin metathesis.[9][10] RCM is a favorite among organic chemists due to its synthetic utility in the formation of rings, which were previously difficult to access efficiently, and broad substrate scope.[11] Since the only major by-product is ethylene, these reactions may also be considered atom economic, an increasingly important concern in the development of green chemistry.[7]
There are several reviews published on ring-closing metathesis.[2][3][12][13]
History
The first example of ring-closing metathesis was reported by Dider Villemin in 1980 when he synthesized an Exaltolide precursor using a WCl6/Me4Sn catalyzed metathesis cylcization in 60-65% yield depending on ring size (A).[8] In the following months, Jiro Tsuji reported a similar metathesis reaction describing the preparation of a macrolide catalyzed by WCl6 and dimethyltitanocene (Cp2TiMe2) in a modest 17.9% yield (B).[14] Tsuji describes the olefin metathesis reaction as “…potentially useful in organic synthesis” and addresses the need for the development of a more versatile catalyst to tolerate various functional groups.

In 1987, Siegfried Warwel and Hans Kaitker published a synthesis of symmetric macrocycles through a cross-metathesis dimerization of starting cycloolefins to afford C14, C18, and C20 dienes in 58-74% yield, as well as C16 in 30% yield, using Re2O7 on Al2O3 and Me4Sn for catalyst activation.[15]

After a decade since its initial discovery, Grubbs and Fu published two influential reports in 1992 detailing the synthesis of O- and N- heterocycles via RCM utilizing Schrock’s molybdenum alkylidene catalysts, which had proven more robust and functional group tolerant than the tungsten chloride catalysts.[16][17] The synthetic route allowed access to dihydropyrans in high yield (89-93%) from readily available starting materials.[16] In addition, synthesis of substituted pyrrolines, tetrahydropyridines, and amides were illustrated in modest to high yield (73-89% ).[17] The driving force for the cylcization reaction was attributed to entropic favorability by forming two molecules per one molecule of starting material. The loss of the second molecule, ethylene, a highly volatile gas, drives the reaction in the forward direction according to Le Châtelier's principle.[16]

In 1993, Grubbs and others not only published a report on carbocycle synthesis using a molybdenum catalyst,[18] but also detailed the initial use of a novel ruthenium carbene complex for metathesis reactions, which later became a popular catalyst due to its extraordinary utility. The ruthenium catalysts are not sensitive to air and moisture, unlike the molybdenum catalysts.[19] The ruthenium catalysts, known better as the Grubbs Catalysts, as well as molybdenum catalysts, or Schrock’s Catalysts, are still used today for many metathesis reactions, including RCM. Overall, it was shown that metal-catalyzed RCM reactions were very effective in C-C bond forming reactions, and would prove of great importance in organic synthesis, chemical biology, materials science, and various other fields to access a wide variety of unsaturated and highly functionalized cyclic analogues.[2][3]
Mechanism
General Mechanism
The mechanism for transition metal-catalyzed olefin metathesis has been widely researched over the past forty years.[20] RCM undergoes a similar mechanistic pathway as other olefin metathesis reactions, such as cross metathesis (CM), ring-opening metathesis polymerization (ROMP), and acyclic diene metathesis (ADMET).[21] Since all steps in the catalytic cycle are considered reversible, it is possible for some of these other pathways to intersect with RCM depending on the reaction conditions and substrates.[12] In 1971, Chauvin proposed the formation of a metallacyclobutane intermediate through a [2+2] cycloaddition[21][22] which then cycloreverts to either yield the same alkene and catalytic species (a nonproductive pathway), or produce a new catalytic species and an alkylidene (a productive pathway).[23] This mechanism has become widely accepted among chemists and serves as the model for the RCM mechanism.[24]

Initiation occurs through substitution of the catalyst’s alkene ligand with substrate. This process occurs via formation of a new alkylidene through one round of [2+2] cycloaddition and cycloreversion. Association and dissociation of a phosphine ligand also occurs in the case of Grubbs catalysts.[25] In an RCM reaction, the alkylidene undergoes an intramolecular [2+2] cycloaddition with the second reactive terminal alkene on the same molecule, rather than an intermolecular addition of a second molecule of starting material, a common competing side reaction which may lead to polymerization[26] Cycloreversion of the metallacyclobutane intermediate forms the desired RCM product along with a [M]=CH2, or alkylidene, species which reenters the catalytic cycle. While the loss of volatile ethylene is a driving force for RCM,[24] it is also generated by competing metathesis reactions and therefore cannot be considered the only driving force of the reaction.[2]
Thermodynamics
The reaction can be under kinetic or thermodynamic control depending on the exact reaction conditions, catalyst, and substrate. Common rings, 5- through 7-membered cycloalkenes, have a high tendency for formation and are often under greater thermodynamic control due to the enthalpic favorability of the cyclic products, as shown by Illuminati and Mandolini on the formation of lactone rings.[27] Smaller rings, between 5 and 8 atoms, are more thermodynamically favored over medium to large rings due to lower ring strain. Ring strain arises from abnormal bond angles resulting in a higher heat of combustion relative to the linear counterpart.[27] If the RCM product contains a strained olefin, polymerization becomes more preferable through ring-opening metathesis polymerization of the newly formed olefin.[28] Medium rings in particular have greater ring strain, in part due to greater transannular interactions from opposing sides of the ring, but also the inability to orient the molecule in such a way to prevent penalizing gauche interactions.[27][29] RCM may be considered to have a kinetic bias if the products cannot reenter the catalytic cycle or interconvert through an equilibrium. A kinetic product distribution could lead to mostly RCM products or may lead to oligomers and polymers, which are most often disfavored.[2]

Equilibrium
With the advent of more reactive catalysts, equilibrium RCM is observed quite often which may lead to a greater product distribution. The mechanism can be expanded to include the various competing equilibrium reactions as well as indicate where various side-products are formed along the reaction pathway, such as oligomers.[30]
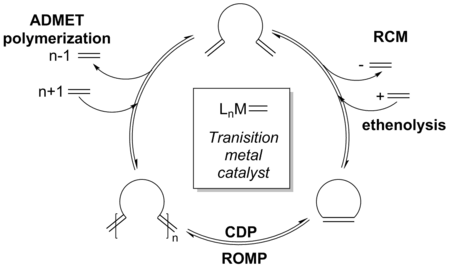
Although the reaction is still under thermodynamic control, an initial kinetic product, which may be dimerization or oligomerization of the starting material, is formed at the onset of the reaction as a result of higher catalyst reactivity. Increased catalyst activity also allows for the olefin products to reenter the catalytic cycle via non-terminal alkene addition onto the catalyst.[2][31][32] Due to additional reactivity in strained olefins, an equilibrium distribution of products is observed; however, this equilibrium can be perturbed through a variety of techniques to overturn the product ratios in favor of the desired RCM product.[33][34]

Since the probability for reactive groups on the same molecule to encounter each other is inversely proportional to the ring size, the necessary intramolecular cycloaddition becomes increasingly difficult as ring size increases. This relationship means that the RCM of large rings is often performed under high dilution (0.05 - 100 mM) (A) [2][35] to reduce intermolecular reactions; while the RCM of common rings can be performed at greater concentrations, even neat in rare cases.[36][37] The equilibrium reaction can be driven to the desired thermodynamic products by increasing temperature (B), to decrease viscosity of the reaction mixture and therefore increase thermal motion, as well as increasing or decreasing reaction time (C).[30][38]

Catalyst choice (D) has also been shown to be critical in controlling product formation. A few of the catalyts commonly used in ring-closing metathesis are shown below.[11][39][40][41]

Reaction Scope
Alkene Substrate
Ring-closing Metathesis has shown utility in the synthesis of 5-30 membered rings,[42] polycycles, and heterocycles containing atoms such as N, O, S, P, and even Si.[2][3][43][44] Due to the functional group tolerance of modern RCM reactions, the synthesis of structurally complex compounds containing a range of functional groups such as epoxides, ketones, alcohols, ethers, amines, amides, and many others can be achieved more easily than previous methods. Oxygen and nitrogen heterocycles dominate due to their abundance in natural products and pharmaceuticals. Some examples are shown below (the red alkene indicates C-C bond formed through RCM).[3]

In addition to terminal alkenes, tri- and tetrasubstituted alkenes have been used in RCM reactions to afford substituted cyclic olefin products.[32] Ring-closing metathesis has also been used to cyclize rings containing an alkyne to produce a new terminal alkene, or even undergo a second cyclization to form bicycles. This type of reaction is more formally known as enyne ring-closing metathesis.[7][45]

E/Z Selectivity
In RCM reactions, two possible geometric isomers, either E- or Z-isomer, may be formed. Stereoselectivity is dependent on the catalyst, ring strain, and starting diene. In smaller rings, Z-isomers predominate as the more stable product reflecting ring-strain minimization.[46] In macrocycles, the E-isomer is often obtained as a result of the thermodynamic bias in RCM reactions as E-isomers are more stable compared to Z-isomers. As a general trend, ruthenium NHC (N-heterocyclic carbene) catalysts favor E selectivity to form the trans isomer. This in part due to the steric clash between the substituents, which adopt a trans configuration as the most stable conformation in the metallacyclobutane intermediate, to form the E-isomer.[21] The synthesis of stereopure Z- isomers were previously achieved via ring-closing alkyne metathesis. However, in 2013 Grubbs reported the use of a chelating ruthenium catalyst to afford Z macrocycles in high selectivity. The selectivity is attributed to the increased steric clash between the catalyst ligands and the metallacyclobutane intermediate that is formed. The increased steric interactions in the transition state lead to the Z olefin rather than the E olefin, because the transition state required to form the E- isomer is highly disfavored.[47]
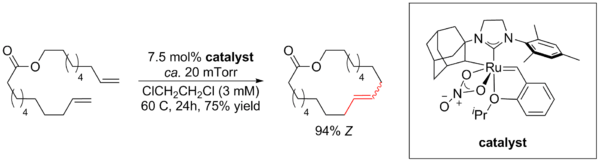
Cocatalyst
Additives are also used to overturn conformational preferences, increase reaction concentration, and chelate highly polar groups, such as esters or amides, which can bind to the catalyst.[2] Titanium isopropoxide (Ti(OiPr)4) is commonly used to chelate polar groups to prevent catalyst poisoning and in the case of an ester, the titanium Lewis acid binds the carbonyl oxygen. Once the oxygen is chelated with the titanium it can no longer bind to the ruthenium metal of the catalyst, which would result in catalyst deactivation. This also allows the reaction to be run at a higher effective concentration without dimerization of starting material.[48]

Another classic example is the use of a bulky Lewis acid to form the E-isomer of an ester over the preferred Z-isomer for cyclolactonization of medium rings. In one study, the addition of aluminum tris(2,6-diphenylphenoxide) (ATPH) was added to form a 7-membered lactone. The aluminum metal binds with the carbonyl oxygen forcing the bulky diphenylphenoxide groups in close proximity to the ester compound. As a result, the ester adopts the E-isomer to minimize penalizing steric interactions. Without the Lewis acid, only the 14-membered dimer ring was observed.[49]

By orienting the molecule in such a way that the two reactive alkenes are in close proximity, the risk of intermolecular cross-metathesis is minimized.
Limitations
Many metathesis reactions with ruthenium catalysts are hampered by unwanted isomerization of the newly formed double bond, and it is believed that ruthenium hydrides that form as a side reaction are responsible. In one study [50] it was found that isomerization is suppressed in the RCM reaction of diallyl ether with specific additives capable of removing these hydrides. Without an additive, the reaction product is 2,3-dihydrofuran and not the expected 2,5-dihydrofuran (together with the formation of ethylene gas). Radical scavengers, such as TEMPO or phenol, do not suppress isomerization; however, additives such as 1,4-benzoquinone or acetic acid successfully prevent unwanted isomerization. Both additives are able to oxidize the ruthenium hydrides which may explain their behavior.

Another common problem associated with RCM is the risk of catalyst degradation due to the high dilution required for some cyclizations. High dilution is also a limiting factor in industrial applications due to the large amount of waste generated from large-scale reactions at a low concentration.[2] Efforts have been made to increase reaction concentration without compromising selectivity.[51]
Synthetic Applications
Ring-closing metathesis has been used historically in numerous organic syntheses and continues to be used today in the synthesis of a variety of compounds. The following examples are only representative of the broad utility of RCM, as there are numerous possibilities. For additional examples see the many review articles.[2][3][13][42]
Ring-closing metathesis is important in total synthesis. One example is its use in the formation of the 12-membered ring in the synthesis of the naturally occurring cyclophane floresolide. Floresolide B was isolated from an ascidian of the genus Apidium and showed cytotoxicity against KB tumor cells. In 2005, K. C. Nicolaou and others completed a synthesis of both isomers through late-stage ring-closing metathesis using the 2nd Generation Grubbs catalyst to afford a mixture of E- and Z- isomers (1:3 E/Z) in 89% yield. Although one prochiral center is present the product is racemic. Floresolide is an atropisomer as the new ring forms (due to steric constraints in the transition state) passing through the front of the carbonyl group in and not the back. The carbonyl group then locks the ring permanently in place. The E/Z isomers were then separated and then the phenol nitrobenzoate protective group was removed in the final step by potassium carbonate to yield the final product and the unnatural Z-isomer.[52]

In 1995, Robert Grubbs and others highlighted the stereoselectivity possible with RCM. The group synthesized a diene with an internal hydrogen bond forming a β-turn. The hydrogen bond stabilized the macrocycle precursor placing both dienes in close proximity, primed for metathesis. After subjecting a mixture of diastereomers to the reaction conditions, only one diastereomer of the olefin β-turn was obtained. The experiment was then repeated with (S,S,S) and (R,S,R) peptides. Only the (S,S,S) diastereomer was reactive illustrating the configuration needed for ring-closing to be possible. It is also interesting that the olefin product’s absolute configuration mimics that of Balaram’s disulfide peptide.[53]

The ring strain in 8-11 atom rings has proven to be challenging for RCM; however, there are many cases where these cyclic systems have been synthesized.[3] In 1997, Fürstner reported a facile synthesis to access jasmine ketolactone (E/Z) through a final RCM step. At the time, no previous 10-membered ring had been formed through RCM, and previous syntheses were often lengthy, involving a macrolactonization to form the decanolide. By adding the diene and catalyst over a 12 hour period to refluxing toluene, Fürstner was able to avoid oligomerization and obtain both E/Z isomers in 88% yield. It is interesting to note that CH2Cl2 favored the formation of the Z-isomer in 1:2.5 (E/Z) ratio, whereas, toluene only afforded a 1:1.4 (E/Z) mixture.[54]

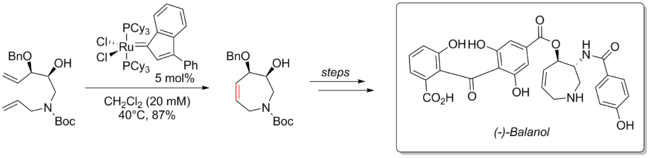
In 2000, Alois Fürstner reported an eight step synthesis to access (−)-balanol using RCM to form a 7-member heterocycle intermediate. Balanol is a metabolite isolated from erticiullium balanoides and shows inhibitory action towards protein kinase C (PKC). In the ring closing metathesis step, a ruthenium indenylidene complex was used as the precatalyst to afford the desired 7-member ring in 87% yield.[55]
In 2002, Stephen F. Martin and others reported the 24-step synthesis of manzamine A with two ring-closing metathesis steps to access the polycyclic alkaloid.[56] The natural product was isolated from marine sponges off the coast of Okinawa. Manzamine is a good target due to its potential as an antitumor compound. The first RCM step was to form the 13-member D ring as solely the Z-isomer in 67% yield, a unique contrast to the usual favored E-isomer of metathesis. After further transformations, the second RCM was used to form the 8-member E ring in 26% yield using stoichiometric 1st Generation Grubbs catalyst. The synthesis highlights the ability for functional group tolerance metathesis reactions as well as the ability to access complex molecules of varying ring sizes.[56]

In 2003, Danishefsky and others reported the total synthesis of (+)-migrastatin, a macrolide isoalated from Streptomyces which inhibited tumor cell migration.[57] The macrolide contains a 14-member heterocycle that was formed through RCM. The metathesis reaction yielded the protected migrastatin in 70% yield as only the (E,E,Z) isomer. It is reported that this selectivity arises from the preference for the ruthenium catalyst to add to the less hindered olefin first then cyclize to the most accessible olefin. The final deprotection of the silyl ether yielded (+)-migrastatin.[57]

Overall, ring-closing metathesis is a highly useful reaction to readily obtain cyclic compounds of varying size and chemical makeup; however, it does have some limitations such as high dilution, selectivity, and unwanted isomerization.
See also
- Olefin Metathesis
- Ring-opening metathesis polymerization
- Alkane metathesis
- Alkyne metathesis
- Enyne metathesis
References
- ↑ Carey, F. A.; Sunburg, R. J. Reactions Involving Transition Metals. Advanced Organic Chemistry: Reaction and Synthesis, 5th Ed.; Part B; Springer: New York, 2010, pp. 761-767.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 2.9 2.10 Monfette, S.; Fogg, D. E. (2009). "Equilibrium Ring-Closing Metathesis". Chem. Rev. 109 (8): 3783-3816. doi: 10.1021/cr800541y.
- ↑ 3.0 3.1 3.2 3.3 3.4 3.5 3.6 Deiters, A.; Martin, S. F. (2004). “Synthesis of Oxygen- and Nitrogen-Containing Heterocycles by Ring-Closing Metathesis”. Chem. Rev. 104 (5): 2199-2238. doi: 10.1021/cr0200872.
- ↑ Cain, M. F.; Forrest, W. P.; Peryshkov, R. V.; Schrock, R. R. Muller, P. (2013). “Synthesis of a TREN in Which the Aryl Substituents are Part of a 45 Atom Macrocycle”. J. Am. Chem. Soc. 135 (41): 15338-15341. doi: 10.1021/ja408964g.
- ↑ Dasgupta, S.; Wu, J. (2011). “Template-directed synthesis of kinetically and thermodynamically stable molecular necklace using ring closing metathesis”. Org. Biomol. Chem. 9: 3504-3515. doi: 10.1039/c0ob01034k
- ↑ Song, K. H.; Kang, S. O.; Ko, (2007). “Template Synthesis of a Huge Macrocycle by Olefin Metathesis Using Easily Accessible [Pt(PEt3)2] Templates”. Chem. Eur. J. 13 (18): 5129–5134. doi: 10.1002/chem.200700213.
- ↑ 7.0 7.1 7.2 Schmalz, H.-G. (1995). “Catalytic Ring-Closing Metathesis : A New, Powerful Technique for Carbon- Carbon Coupling in Organic Synthesis”. Angew. Chem. Int. Ed. Engl. 34 (17): 1833-1836. doi: 10.1002/anie.199518331.
- ↑ 8.0 8.1 Villemin, D. (1980). “Synthese de Macrolides par Metathese”. Tetrahedron Lett. 21 (18): 1715-1718. doi:10.1016/S0040-4039(00)77818-X.
- ↑ Grubbs, R. H. (2006). “Olefin-Metathesis Catalysts for the Preparation of Molecules and Materials (Nobel Lecture)”. Angew. Chem., Int. Ed. 45 (23): 3760–3765. doi: 10.1002/anie.200600680.
- ↑ Schrock, R. R. (2006). “Multiple Metal–Carbon Bonds for Catalytic Metathesis Reactions (Nobel Lecture)”. Angew. Chem., Int. Ed. 45 (23), 3748-3759. doi: 10.1002/anie.200600085.
- ↑ 11.0 11.1 Trnka, T. M.; Grubbs, R. H. (2001). “The Development of L2X2Ru=CHR Olefin Metathesis Catalysts: An Organometallic Success Story”. Acc. Chem. Res. 34 (1):18-29. doi: 10.1021/ar000114f.
- ↑ 12.0 12.1 Furstner, A. (2000). “Olefin Metathesis and Beyond”. Angew. Chem., Int. Ed. 39 (17): 3012-3043. doi: 10.1002/1521-3773(20000901)39:17<3012::AID-ANIE3012>3.0.CO;2-G.
- ↑ 13.0 13.1 Gradillas, A.; Perez-Castells, J. (2006). “Macrocyclization by Ring-Closing Metathesis in the Total Synthesis of Natural Products: Reaction Conditions and Limitations”. Angew. Chem., Int. Ed. 45: 6086-6101. doi: 10.1002/anie.200600641.
- ↑ Tsuji, J.; Hashiguchi, S. (1980). “Application of Olefin Metathesis to Organic Synthesis. Syntheses of Civetone and Macrolides”. Tetrahedron Lett. 21 (31): 2955-2958. doi: 10.1016/0040-4039(80)88007-5.
- ↑ Warwel, S.; Katker, H. (1987). “Eine einfache Synthese makrocyclischer Kohlenwasserstoffe durch Metathese von Cyclooflefinen”. Synthesis. 935-937.
- ↑ 16.0 16.1 16.2 Fu, G. C.; Grubbs, R. H. (1992). “The Application of Catalytic Ring-Closing Olefin Metathesis to the Synthesis of Unsaturated Oxygen Heterocycles”. J. Am. Chem. Soc. 114 (13): 5426-5427. doi: 10.1021/ja00039a065.
- ↑ 17.0 17.1 Fu, G. C.; Grubbs, R. H. (1992).“Synthesis of Nitrogen Heterocycles via Catalytic RingClosing Metathesis of Dienes”. J. Am. Chem. Soc. 114 (18): 7324-7325. doi: 10.1021/ja00044a070.
- ↑ Fu, G. C.; Grubbs, R. H. (1993). “Synthesis of cycloalkenes via alkylidene-mediated olefin metathesis and carbonyl olefination”. J. Am. Chem. Soc. 115 (9): 3800-3801. doi: 10.1021/ja00062a066.
- ↑ Fu, G. C.; Nguyen, S. T.; Grubbs, R. H. (1993). “Catalytic Ring-Closing Metathesis of Functionalized Dienes by a Ruthenium Carbene Complex”. J. Am. Chem. Soc. 115 (21): 9856-9857. doi: 10.1021/ja00074a085.
- ↑ Chauvin, Y. (2006). “Olefin Metathesis: The Early Days (Nobel Lecture). Angew. Chem., Int. Ed. 43 (23): 3740-3747”. doi: 10.1002/anie.200601234.
- ↑ 21.0 21.1 21.2 Crabtree, R. H. Applications. The Organometallic Chemistry of the Transition Metals, 6th Ed.; John Wiley & Sons, Inc.: New Jersey, 2014, pp.318-322.
- ↑ Herisson, J-L.; Chauvin, Y. (1971). “Catalyse de transformation des olefines par les complexes du tungsten”. Makromolekulare Chemie. 141 (1): 161-176. doi: 10.1002/macp.1971.021410112.
- ↑ Stewart, I. C.; Keitz, B. K.; Kuhn, K. M.; Thomas, R. M.; Grubbs, R. H. (2010). “Nonproductive Events in Ring-Closing Metathesis Using Ruthenium Catalysts”. J. Am. Chem. Soc. 132 (25), 8534-8535. doi: 10.1021/ja1029045.
- ↑ 24.0 24.1 Grossman, R. B. Transition-Metal-Catalyzed & -Mediated Reactions. The Art of Writing Reasonable Organic Reaction Mechanisms, 2nd Ed.; Springer: New York, 2003, pp. 324-325.
- ↑ Ansyln, E. V.; Dougherty, D. A. Organotransition Metal Reaction Mechanisms and Catalysts. Modern Physical Organic Chemistry, Murdzek, J., Ed. University Science Books, 2006, pp. 745-746.
- ↑ Lee, C. W.; Grubbs, R. H. (2001). “Formation of Macrocycles via Ring-Closing Olefin Metathesis”. J. Org. Chem. 66 (21):7155-7158. doi: 10.1021/jo0158480.
- ↑ 27.0 27.1 27.2 Illuminati, G.; Mandolini, L. (1981). “Ring Closure Reactions of Bifunctional Chain Molecules”. Acc. Chem. Res. 14 (5): 95-102. doi: 10.1021/ar00064a001.
- ↑ http://faculty.chemistry.harvard.edu/files/myers/files/31-the_olefin_metathesis_reaction.pdf
- ↑ Anslyn, E. V.; Dougherty, D. A. Strain and Stability. Modern Physical Organic Chemistry, Murdzek, J., Ed. University Science Books, 2006, pp. 107-111.
- ↑ 30.0 30.1 Conrad, J. C.; Eelman, M. D.; Duarte Silva, J. A.; Monfette, S.; Parnas, H. H.; Snelgrove, J. L.; Fogg, D. E. (2007). “Oligomers as Intermediates in Ring-Closing Metathesis”. J. Am. Chem. Soc. 129 (5): 1024-1025. doi: 10.1021/ja067531t.
- ↑ Hocker, H. (1991). “Metathesis polymerization - stepwise or chain growth reaction?”. J. Mol. Catal. 65 (1-2): 95–99. doi: 10.1016/0304-5102(91)85086-H.
- ↑ 32.0 32.1 Stewart, I. C.; Ung, T.; Pletnev, A. A.; Berlin, J. B.; Grubbs, R. H.; Schrodi, Y. (2007). “Highly Efficient Ruthenium Catalysts for the Formation of Tetrasubstituted Olefins via Ring-Closing Metathesis”. Org. Lett. 9 (8): 1589-1592. doi: 10.1021/ol0705144.
- ↑ Forbes, M. D. E.; Patton, J. T.; Myers, T. L.; Maynard, H. D.; Smith, Jr. D. W.; Schulz, G. R.; Wagener, K. B. (1992). Solvent-Free Cyclization of Linear Dienes Using Olefin Metathesis and the Thorpe lngold Effect". J. Am. Chem. Soc. 114 (27): 10978-10980. doi: 10.1021/ja00053a054.
- ↑ Yamamoto, K.; Biswas, K.; Gaul, C.; Danishefsky, S. J. (2003). “Effects of temperature and concentration in some ring closing metathesis reactions”. Tetrahedron Lett. 44 (16): 3297–3299. doi: 10.1016/S0040-4039(03)00618-X.
- ↑ Arakawa, K.; Eguchi, T.; Kakinuma, K. (1998). “An Olefin Metathesis Approach to 36- and 72-Membered Archaeal Macrocyclic Membrane Lipids”. J. Org. Chem. 63 (14): 4741–4745. doi: 10.1021/jo980472k.
- ↑ Kuhn, K. M.; Champagne, T. M.; Hong, S. H.; Wei, W-H.; Nickel, A.; Lee, C. W.; Virgil, S. C.; Grubbs, R. H.; Pederson, R. L. (2010). “Low Catalyst Loadings in Olefin Metathesis: Synthesis of Nitrogen Heterocycles by Ring-Closing Metathesis”. Org. Lett. 12 (5): 984-987. doi: 10.1021/ol9029808.
- ↑ Bach, T.; Lemarchand, A. (2002). “Synthesis of Ansa-Bridged Macrocyclic Lactams Related to the Antitumor Antibiotic Geldanamycin by Ring Closing Metathesis”. Synlett. 8: 1302-1304. doi: 10.1055/s-2002-32958.
- ↑ Crimmins, M. T.; Brown, B. H. (2004). “An Intramolecular Diels-Alder Approach to the Eunicelins: Enantioselective Total Synthesis of Ophirin B”. J. Am. Chem. Soc. 126 (33): 10264–10266. doi: 10.1021/ja046574b.
- ↑ Xu, Z.; Johannes, C. W.; Houri, A. F.; La, D. S.; Cogan, D. A.; Hofilena, G. E.; Hoveyda, A. H. (1997). “Applications of Zr-Catalyzed Carbomagnesation and Mo-Catalyzed Macrocyclic Ring Closing Metathesis in Asymmetric Synthesis. Enantioselective Total Synthesis of Sch 38516 (Fluvirucin B1)”. J. Am. Chem. Soc. 119 (43): 10302–10316. doi: 10.1021/ja972191k.
- ↑ Furstner, A.; Thiel, O. R.; Ackermann, L. (2001). “Exploiting the Reversibility of Olefin Metathesis. Syntheses of Macrocyclic Trisubstituted Alkenes and (R,R)-(−)-Pyrenophorin”. Org. Lett. 3 (3): 449–451. doi: 10.1021/ol0069554.
- ↑ Furstner, A.; Thiel, O. R.; Kindler, N.; Bartkowska, B. (2000). “Total Syntheses of (S)-(−)-Zearalenone and Lasiodiplodin Reveal Superior Metathesis Activity of Ruthenium Carbene Complexes with Imidazol-2-ylidene Ligands”. J. Org. Chem. 65 (23): 7990–7995. doi: 10.1021/jo0009999.
- ↑ 42.0 42.1 http://www.organic-chemistry.org/namedreactions/ring-closing-metathesis.shtm
- ↑ Harvey, J. S.; Malcolmson, S. J.; Dunne, K. S.; Meek, S. J.; Thompson, A. L.; Schrock, R. R.; Hoveyda, A. H.; Gouverneur, V. (2008). “Enantioselective Synthesis of P-Stereogenic phosphinates and Phosphine Oxides by Molybdenum-Catalyzed Asymmetric Ring-Closing Metathesis”. Angew. Chem., Int. Ed. 48 (4): 762-766. doi: 10.1002/anie.200805066.
- ↑ Kiely, A. F.; Jernelius, J. A.; Schrock, R. R.; Hoveyda, A. H. (2002). “Enantioselective Synthesis of Medium-Ring Heterocycles, Tertiary Ethers, and Tertiary Alcohols by Mo-Catalyzed Ring-Closing Metathesis”. J. Am. Chem. Soc. 124 (12): 2868-2869. doi: 10.1021/ja012679s.
- ↑ Kim, S.-H.; Bowden, N.; Grubbs, R. H. (1994). “Catalytic Ring Closing Metathesis of Dienynes: Construction of Fused Bicyclic Rings”. J. Am. Chem. Soc. 116 (23): 10801-10802. doi: 10.1021/ja00102a062.
- ↑ Anslyn, E. V.; Dougherty, D. A. Strain and Stability. Modern Physical Organic Chemistry, Murdzek, J., Ed. University Science Books, 2006, pp. 110-114.
- ↑ Marx, V. M.; Keitz, B. K.; Grubbs, R. H. (2013). “Stereoselective Access to Z and E Macrocycles by Ruthenium-Catalyzed Z‑Selective Ring-Closing Metathesis and Ethenolysis”. J. Am. Chem. Soc. 135 (1): 94-97. doi: 10.1021/ja311241q.
- ↑ Mitchell, L.; Parkinson, J. A.; Percy, J. M.; Singh, K. (2008). “Selected Substituent Effects on the Rate and Efficiency of Formation of an Eight-Membered Ring by RCM”. J. Org. Chem. 73 (6): 2389–2395. doi: 10.1021/jo702726b.
- ↑ Pentzer, E. B.; Gadzikwa, T.; Nguyen, S. T. (2008). “Substrate Encapsulation: An Efficient Strategy for the RCM Synthesis of Unsaturated ϵ-Lactones”. Org. Lett. 10 (24): 5613-5615. doi: 10.1021/ol8022227.
- ↑ Hong, S. H.; Sanders, D. P.; Lee, C. W.; Grubbs, R. H. (2005). "Prevention of Undesirable Isomerization during Olefin Metathesis". J. Am. Chem. Soc. 127 (49): 17160–17161. doi:10.1021/ja052939w. PubMed.
- ↑ Raymond, M.; Holtz-Mulholland, M.; Collins, S. K. (2014). “Macrocyclic Olefin Metathesis at High Concentrations by Using a Phase-Separation Strategy”. Chem. Eur. J. 20 (4): 12763-12767. doi: 10.1002/chem.201404202.
- ↑ Nicolaou, K. C.; Xu, H. (2006). “Total synthesis of floresolide B and 6,7-Z-floresolide B”. Chem. Commun. 6: 600-602. doi: 10.1039/B517385J.
- ↑ Miller, S. J.; Grubbs, R. H. (1995). “Synthesis of Conformationally Restricted Amino Acids and Peptides Employing Olefin Metathesis”. J. Am. Chem. Soc. 117 (21), 5855-5856. doi: 10.1021/ja00126a027.
- ↑ Furstner, A.; Muller, T. (1997). “The First Synthesis of a 10-Membered Ring by Olefin Metathesis: Jasmine Ketolactone”. Syn. Lett. 8: 1010-1012. doi: 10.1055/s-1997-930.
- ↑ Furstner, A.; Thiel, O. R. (2000). “Formal Total Synthesis of (−)-Balanol: Concise Approach to the Hexahydroazepine Segment Based on RCM” . J. Org. Chem. 65 (6): 1738-1742. doi: 10.1021/jo991611g.
- ↑ 56.0 56.1 Humphrey, J. H.; Liao, Y.; Ali, A.; Rein, T.; Wong, Y.-L.; Chen, H.-J.; Courtney, A. K.; Martin, S. F. (2002). “Enantioselective Total Syntheses of Manzamine A and Related Alkaloids”. J. Am. Chem. Soc. 124 (29): 8584-8592. doi: 10.1021/ja0202964.
- ↑ 57.0 57.1 Gaul, C.; Njardarson, J. T.; Danishefsky, S. J. (2003). “The Total Synthesis of (+)-Migrastatin”. J. Am. Chem. Soc. 125 (20): 6042-6043. doi: 10.1021/ja0349103.
External links
- Ring-Closing Metathesis at organic-chemistry.org
- Sigma-Aldrich Ring-Closing Metathesis at sigmaaldrich.com
- The Olefin Metathesis Reaction Andrew Myers’ Group Notes