R-factor (crystallography)
In crystallography, the R-factor (sometimes called residual factor or reliability factor or the R-value or RWork) is a measure of the agreement between the crystallographic model and the experimental X-ray diffraction data. In other words, it is a measure of how well the refined structure predicts the observed data.[1] The value is also sometimes called the discrepancy index, as it mathematically describes the difference between the experimental observations and the ideal calculated values.[2] It is defined by the following equation:
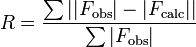
where F is the so-called structure factor and the sum extends over all the reflections measured and their calculated counterparts respectively. The structure factor is closely related to the intensity of the reflection it describes:
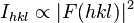
The minimum possible value is zero, indicating perfect agreement between experimental observations and the structure factors predicted from the model. There is no theoretical maximum, but in practice, values are considerably less than one even for poor models, provided the model includes a suitable scale factor. Random experimental errors in the data contribute to 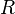 even for a perfect model, and these have more leverage when the data are weak or few, such as for a low-resolution data set. Model inadequancies such as wrong or missing parts and unmodeled disorder are the other main contributors to , making it useful to assess the progress and final result of a crystallographic model refinement. For large molecules, the R-factor usually ranges between 0.6 (when computed for a random model and against an experimental data set) and 0.2 (for example for a well refined macro-molecular model at a resolution of 2.5 Ångström). Small molecules (up to ca. 1000 atoms) usually form better-ordered crystals than large molecules, for which it is possible to attain lower R-factors. In the Cambridge Structural Database of small-molecule structures, more than 95% of the 500,000+ crystals have an R-factor lower than 0.15, and 9.5% have an R-factor lower than 0.03.
even for a perfect model, and these have more leverage when the data are weak or few, such as for a low-resolution data set. Model inadequancies such as wrong or missing parts and unmodeled disorder are the other main contributors to , making it useful to assess the progress and final result of a crystallographic model refinement. For large molecules, the R-factor usually ranges between 0.6 (when computed for a random model and against an experimental data set) and 0.2 (for example for a well refined macro-molecular model at a resolution of 2.5 Ångström). Small molecules (up to ca. 1000 atoms) usually form better-ordered crystals than large molecules, for which it is possible to attain lower R-factors. In the Cambridge Structural Database of small-molecule structures, more than 95% of the 500,000+ crystals have an R-factor lower than 0.15, and 9.5% have an R-factor lower than 0.03.
Crystallographers also use the Free R-Factor (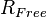 ) [3] to assess possible overmodelling of the data. is computed according to the same formula given above, but on a small, random sample of data that are set aside for the purpose and never included in the refinement. will always be greater than because the model is not fitted to the reflections that contribute to , but the two statistics should be similar because a correct model should predict all the data with uniform accuracy. If the two statistics differ significantly then that indicates the model has been over-parameterized, so that to some extent it predicts not the ideal error-free data for the correct model, but rather the error-afflicted data actually observed.
) [3] to assess possible overmodelling of the data. is computed according to the same formula given above, but on a small, random sample of data that are set aside for the purpose and never included in the refinement. will always be greater than because the model is not fitted to the reflections that contribute to , but the two statistics should be similar because a correct model should predict all the data with uniform accuracy. If the two statistics differ significantly then that indicates the model has been over-parameterized, so that to some extent it predicts not the ideal error-free data for the correct model, but rather the error-afflicted data actually observed.
The quantities 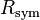 and
and 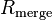 are similarly used to describe the internal agreement of measurements in a crystallographic data set.
are similarly used to describe the internal agreement of measurements in a crystallographic data set.
References
- ↑ Morris AL, MacArthur MW, Hutchinson EG, Thornton JM (April 1992). "Stereochemical quality of protein structure coordinates". Proteins 12 (4): 345–64. doi:10.1002/prot.340120407. PMID 1579569.
- ↑ "R factor". International Union of Crystallography. Retrieved 2013-12-13.
- ↑ Brunger AT (January 1992). "Free R value: a novel statistical quantity for assessing the accuracy of crystal structures". Nature 355 (6359): 472–475. Bibcode:1992Natur.355..472B. doi:10.1038/355472a0. PMID 18481394.