Quantitative proteomics
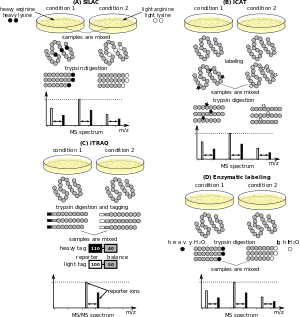
Quantitative proteomics is an analytical chemistry technique for determining the amount of proteins in a sample.[1][2][3] Rather than just providing lists of proteins identified in a certain sample, quantitative proteomics yields information about differences between samples. For example, this approach can be used to compare samples from healthy and diseased patients. The methods for protein identification are identical to those used in general (i.e. qualitative) proteomics, but include quantification as an additional dimension. Quantitative proteomics is mainly performed by two-dimensional gel electrophoresis (2-DE) or mass spectrometry (MS). In contrast to 2-DE, which requires MS for the downstream protein identification, MS technology can identify and quantitate the changes.
Technologies
Mass spectrometry (MS) and two-dimensional gel electrophoresis (2-DE) represent the main technologies for quantitative proteomics with advantages and disadvantages. 2-DE provides information about the protein quantity, charge, and mass of the intact protein. It has limitations for the analysis of proteins larger than 150 kDa or smaller than 5kDa and low solubility proteins. Quantitative MS has higher sensitivity but does not provide information about the intact protein.
Classical 2-DE based on post-electrophoretic dye staining has limitations: at least three technical replicates are required to verify the reproducibility. Difference gel electrophoresis (DIGE) uses fluorescence-based labeling of the proteins prior to separation has increased the precision of quantification as well as the sensitivity in the protein detection. Therefore DIGE represents the current main approach for the 2-DE based study of proteomes.
For quantitative MS, a commonly applied approach is isotope-coded affinity tags (ICAT), which uses two reagents with heavy and light isotopes, respectively, and a biotin affinity tag to modify cysteine containing peptides. This technology has been used to label whole Saccharomyces cerevisiae cells,[4] and, in conjunction with mass spectrometry, helped lay the foundation of quantitative proteomics.
Discovery and targeted proteomics
Strategies to improve the sensitivity and scope of proteomic analysis often require large sample quantities and multi-dimensional fractionation, which sacrifices throughput. Alternatively, efforts to improve the sensitivity and throughput of protein quantification limit the number of peptides that can be monitored per MS run. For this reason, proteomics research is typically divided into two categories: discovery and targeted proteomics. Discovery proteomics optimizes protein identification by spending more time and effort per sample and reducing the number of samples analyzed. In contrast, targeted proteomics strategies limit the number of features that will be monitored and then optimize the chromatography, instrument tuning and acquisition methods to achieve the highest sensitivity and throughput for hundreds or thousands of samples.
Relative and absolute quantitation
Mass spectrometry is not inherently quantitative because of differences in the ionization efficiency and/or detectability of the many peptides in a given sample, which has sparked the development of methods to determine relative and absolute abundance of proteins in samples.[3] The intensity of a peak in a mass spectrum is not a good indicator of the amount of the analyte in the sample, although differences in peak intensity of the same analyte between multiple samples accurately reflect relative differences in its abundance.
Label-free quantification
One approach for relative quantitation is to separately analyze samples by MS and compare the spectra to determine peptide abundance in one sample relative to another, as in Label-free quantification strategies.
Stable isotope labels
An approach for relative quantitation that is more costly and time-consuming, though less sensitive to experimental bias than label-free quantitation, entails labeling the samples with stable isotope labels that allow the mass spectrometer to distinguish between identical proteins in separate samples. One type of label, isotopic tags, consist of stable isotopes incorporated into protein crosslinkers that causes a known mass shift of the labeled protein or peptide in the mass spectrum. Differentially labeled samples are combined and analyzed together, and the differences in the peak intensities of the isotope pairs accurately reflect difference in the abundance of the corresponding proteins.
Absolute proteomic quantitation using isotopic peptides entails spiking known concentrations of synthetic, heavy isotopologues of target peptides into an experimental sample and then performing LC-MS/MS. As with relative quantitation using isotopic labels, peptides of equal chemistry co-elute and are analyzed by MS simultaneously. Unlike relative quantitation, though, the abundance of the target peptide in the experimental sample is compared to that of the heavy peptide and back-calculated to the initial concentration of the standard using a pre-determined standard curve to yield the absolute quantitation of the target peptide.
Relative quantitation methods include isotope-coded affinity tags (ICAT), isobaric labeling (tandem mass tags (TMT) and isobaric tags for relative and absolute quantitation (iTRAQ)), label-free quantification Metal-coded tags (MeCAT), N-terminal labelling, stable isotope labeling with amino acids in cell culture (SILAC), and Terminal amine isotopic labeling of substrates (TAILS).
Absolute quantitation is performed using selected reaction monitoring (SRM).
MeCAT can be used in combination with element mass spectrometry ICP-MS allowing first-time absolute quantification of the metal bound by MeCAT reagent to a protein or biomolecule. Thus it is possible to determine the absolute amount of protein down to attomol range using external calibration by metal standard solution. It is compatible to protein separation by 2D electrophoresis and chromatography in multiplex experiments. Protein identification and relative quantification can be performed by MALDI-MS/MS and ESI-MS/MS.
Mass spectrometers have a limited capacity to detect low-abundance peptides in samples with a high dynamic range. The limited duty cycle of mass spectrometers also restricts the collision rate, resulting in an undersampling[5] Sample preparation protocols represent sources of experimental bias.
See also
References
- ↑ Ong SE, Mann M (2005). "Mass spectrometry-based proteomics turns quantitative". Nature Chemical Biology 1 (5): 252–262. doi:10.1038/nchembio736. PMID 16408053.
- ↑ Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B (October 2007). "Quantitative mass spectrometry in proteomics: a critical review". Anal Bioanal Chem 389 (4): 1017–31. doi:10.1007/s00216-007-1486-6. PMID 17668192.
- ↑ 3.0 3.1 Nikolov M, Schmidt C, Urlaub H (2012). "Quantitative Mass Spectrometry-Based Proteomics: An Overview". Methods in Molecular Biology 893: 85–100. doi:10.1007/978-1-61779-885-6_7. PMID 22665296.
- ↑ Oda Y, Huang K, Cross FR, Cowburn D, Chait BT (June 1999). "Accurate quantitation of protein expression and site-specific phosphorylation". Proc. Natl. Acad. Sci. U.S.A. 96 (12): 6591–6. doi:10.1073/pnas.96.12.6591. PMC 21959. PMID 10359756.
- ↑ Prakash, A. et al. (2007). "Assessing bias in experiment design for large scale mass spectrometry-based quantitative proteomics". Mol Cell Proteomics 389 (10): 1017–31. doi:10.1074/mcp.M600470-MCP200. PMID 17617667.
| ||||||||||