Pulvinone
-Pulvinone_Structural_Formulae.png) | |
| Names | |
|---|---|
| IUPAC name
(E/Z)-5-Phenylmethylene-4-hydroxy-3-phenylfuran-2(5H)-one | |
| Identifiers | |
| 4941-88-2 (E/Z) 78376-10-0 (Z) 100074-80-4 (E) | |
| Properties | |
| Molecular formula |
C17H12O3 |
| Molar mass | 264.28 g·mol−1 |
| Appearance | Yellow needles |
| Melting point | 243 to 247 °C (469 to 477 °F; 516 to 520 K)[1] |
| Insoluble | |
| Related compounds | |
| Related compounds |
Pulvinic acid, Vulpinic acid |
| Except where noted otherwise, data is given for materials in their standard state (at 25 °C (77 °F), 100 kPa) | |
| | |
| Infobox references | |
Pulvinone, an organic compound belonging to the esters, lactones, alcohols and butenolides classes, is a yellow crystalline solid. Although the pulvinone is not a natural product, several naturally occurring hydroxylated derivatives are known. These hydroxylated pulvinones are produced by fungal species, such as the in Europe common Larch Bolete (Boletus elegans, also known as Suillus grevillei), or by moulds such as Aspergillus Terreus.
History
Fungi - such as boleti -, moulds and lichens produce a wide range of pigments made up of one (monomer) or several (oligomers) units of pulvinic acid. In 1831, in the course of a study of the constituents of lichens (Cetraria Vulpina), the French chemist and pharmacist Antoine Bebert discovered a compound named vulpinic acid, the first known naturally occurring methyl ester of pulvinic acid. More details about the structure of this pigment were disclosed in 1860 by the German chemists Franz Möller and Adolph Strecker.[2] While trying to elucidate the structure of vulpinic acid, the German chemist Adolf Spiegel [3][4][5][6] found in 1880 that the vulpinic acid could be saponified to a diacid. He named the resulting diacid pulvinic acid. The German chemist Jacob Volhard[7] elucidated the constitution of pulvinic acid by synthesizing it through the basic hydrolysis of a corresponding dicyanocompound. In the process, he also obtained small amounts of a side-product. One year later Ludwig Claisen and Th. Ewan[8] achieved the synthesis of this side-product and characterized it as the 5-benzylidene-4-hydroxy-3-phenylfuran-2(5H)-one.
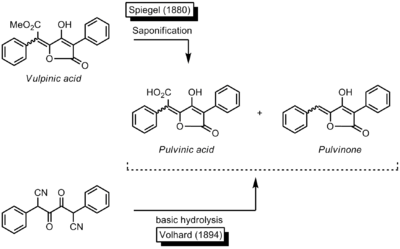
Claisen and Ewan described it as das der Pulvinsäure zu Grunde liegende Lacton (the lactone underlying the structure of pulvinic acid): that was the origin of the name pulvinone.
Natural occurrence
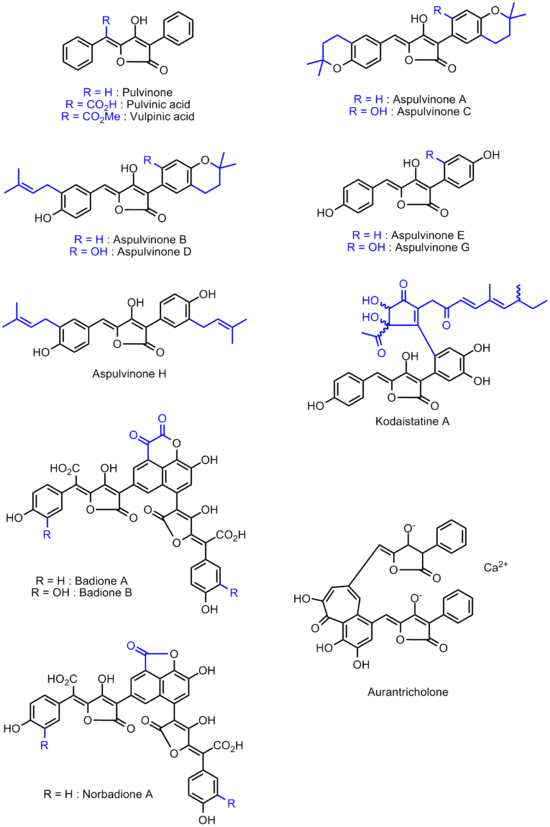
Interestingly, it is only a century after the synthesis of the first pulvinone that the word Pulvinone turned into a collective term. In 1973, Edwards and Gill isolated the first naturally occurring hydroxylated pulvinone derivative.[9] This trihydroxylated pulvinone was found as one of the main pigments responsible for the yellow colour of the stem and caps of the European mushroom Larch Bolete (Boletus elegans, also known as Suillus grevillei). In the very same year 1973, Seto and coworkers also found hydroxylated pulvinones in cultures of the mould Aspergillus terreus.[10][11][12] To insist on their origin - and thereby differentiate them from the hydroxylated pulvinones found in Suillus grevillei - Seto and coworkers named these compounds Aspulvinones.[13][14][15][16] The aspulvinone terminology also incorporates a letter, indicating the order of chromatographic elution of these compounds (hence, the least polar aspulvinone was named Aspulvinone A, the one eluting next Aspulvinone B, etc...).
Like many other yellow pigments in fungi and lichens, pulvinones can be traced back from the pulvinic acid pathway. The pulvinone structural unit is found in a number of natural products. All monomeric (such as pulvinic acid itself, vulpinic acid, comphidic acid, the aspulvinones and the Kodaistatins[17]) or oligomeric (Badiones,[18] Norbadione,[19][20] Aurantricholon[21]) derivatives of the pulvinic acid contain the pulvinone structural element. So far, all naturally occurring pulvinone derivatives were found to be Z-configured.
Pharmacological properties
- Rehse et al.[22][23][24][25] showed the anti-coagulant activity of some pulvinones in rats.
- At the beginning of the 80s, the companies ICI and Smith Kline & French patented a large number of derivatives of the vulpinic acid because of their anti-inflammatory, anti-fever and pain-killing properties. Yet vulpinic acid - as well as many of its derivatives - is a cytotoxic compound. Since pulvinones exhibit a lower cytotoxicity compared to vlupinic acid and its derivatives, Organon investigated the pharmaceutical potential of more than 100 pulvinones.[26] To date, the results of these studies were not fully disclosed.
- In 2005, the Wyeth company patented biphenyl-substituted pulvinones[27][28] due to their promising activity against Gram-positive bacteria, including otherwise resistant bacteria. However, pulvinone-based antibiotics were so far only patented for animal use.
Chemical properties
Pulvinone is a lactone, more precisely an intramolecular ester of the trans-1,4-diphenyl-2,3-dihydroxy-1,3-butadiene-1-carboxylic acid, from which it can be prepared through removal of one equivalent of water:
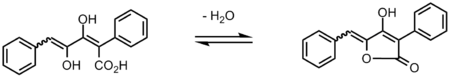
The central 5-membered ring core of pulvinone reveals a 4-hydroxy-butenolide structure. They are essentially found in their enol form, which exhibits acidic properties due to the relative lability of the hydroxylic proton. 4-hydroxy-butenolides such as pulvinones are therefore referred to as tetronic acids, and belong to the larger category of vinylogous acids.
Biosynthesis
The fungal biosynthesis starts from aromatic aminoacids such as phenylalanine and tyrosine; after oxydeamination to the corresponding arylpyruvic acid, the pulvinone skeleton is formed by a sequence of dimerisation, oxidative ring-cleavage and decarboxylation.[29]

Total synthesis
Jacob Volhard was the first to synthesise vulpinic acid, pulvinic acid and pulvinone.[7] To date, 11 total syntheses of pulvinones were reported:
- 1895 by Claisen and Ewan,[8]
- 1975 and 1979 by Knight and Pattenden,[30][31]
- 1979 by Jerris, Wovkulich and Amos B. Smith III,[32]
- 1984 by Ramage et al.,[1]
- 1985 by Campbell et al.,[26]
- 1990 by Gill et al.,[33]
- 1991 by Pattenden, Turvill and Chorlton,[34]
- 2005 by Caufield et al.,[27]
- 2006 by Antane et al.,[28]
- 2007 by Kaczybura and Brückner,[35]
- 2007 by Bernier, Moser and Brückner.[36][37]
See also
Sources
- ↑ 1.0 1.1 Ramage, Robert; Griffiths, Gareth J.; Shutt, Fiona E.; Sweeney, John N. A. (1984). "Dioxolanones as synthetic intermediates. Part 2. Synthesis of tetronic acids and pulvinones". Journal of the Chemical Society, Perkin Transactions 1: 1539. doi:10.1039/P19840001539.
- ↑ Canstatt's Jahresbericht über die Fortschritte in der Pharmacie und verwandte Wissenschaften in allen Ländern, Harvard Universität, Jahrgang 10 (1861).
- ↑ A. Spiegel, Ber. Dtsch. Chem. Ges. 1880, 13, 2, 1629-1635 doi:10.1002/cber.18800130293.
- ↑ A. Spiegel, Ber. Dtsch. Chem. Ges. 1880, 13, 2, 2219-2221 doi:10.1002/cber.188001302237.
- ↑ A. Spiegel, Ber. Dtsch. Chem. Ges. 1881, 14, 1, 873-874 doi:10.1002/cber.188101401183.
- ↑ 6.0 6.1 A. Spiegel, Ber. Dtsch. Chem. Ges. 1881, 14, 2, 1686-1696 doi:10.1002/cber.18810140230.
- ↑ 7.0 7.1 7.2 J. Volhard, Annal. Chem. 1894, 282, 1-21 doi:10.1002/jlac.18942820102.
- ↑ 8.0 8.1 L. Claisen, Th. Ewan, Annal. Chem., 1895, 284, 245-299 doi:10.1002/jlac.18952840302.
- ↑ R. L. Edwards, M. Gill, J. Chem. Soc., Perkin Trans. 1 1973, 1921-1929. doi:10.1039/P19730001921.
- ↑ N. Ojima, S. Takenaka, S. Seto, Phytochemistry (Elsevier) 1973, 12, 2527-2529.
- ↑ N. Ojima, K. Ogura, S. Seto, J. Chem. Soc., Chem. Commun. 1975, 717-718.
- ↑ N. Ojima, S. Takenaka, S. Seto, Phytochemistry (Elsevier) 1975, 14, 573-576.
- ↑ N. Ojima, I. Takahashi, K. Ogura, S. Seto, Tetrahedron Lett. 1976, 17, 1013-1014.
- ↑ I. Takahashi, N. Ojima, K. Ogura, S. Seto, Biochemistry 1978, 17, 2696-2702.
- ↑ M. Kobayashi, N. Ojima, K. Ogura, S. Seto, Chem. Lett. 1979, 579-582.
- ↑ H. Sugiyama, N. Ojima, M. Kobayashi, Y. Senda, J. Ishiyama, S. Seto, Agric. Biol. Chem. 1979, 43, 403-4.
- ↑ L. Vértesy, H.-J. Burger, J. Kenja, M. Knauf, H. Kogler, E. F. Paulus, N. V. S. Ramakrishna, K. H. S. Swamy, E. K. S. Vijayakumar, P. Hammann, J. Antibiot. 2000, 53, 677-686.
- ↑ B. Steffan, W. Steglich, Angew. Chem., Int. Ed. 1984, 23, 6, 445-447. doi:10.1002/anie.198404451.
- ↑ M. Gill, D. A. Lally, Phytochemistry 1985, 24, 1351-1354. doi:10.1016/S0031-9422(00)81131-0.
- ↑ T. Le Gall, C. Mioskowski, B. Amekraz et al. Angew. Chem., Int. Ed. 2003, 42, 11, 1289-1293. doi:10.1002/anie.198404451.
- ↑ D. Klostermeyer, L. Knops, T. Sindlinger, K. Polborn, W. Steglich Eur. J. Org. Chem. 2000, 4, 603-609. doi:10.1002/anie.200390332.
- ↑ K. Rehse, J. Wagenknecht, N. Rietbrock, Arch. Pharm. (Weinheim, Ger.) 1978, 311, 986-991.
- ↑ K. Rehse, U. Emisch, Arch. Pharm. (Weinheim, Ger.) 1982, 315, 1020-1025.
- ↑ K. Rehse, J. Schinke, G. Bochert, Arch. Pharm. (Weinheim, Ger.) 1979, 312, 390-394.
- ↑ K. Rehse, J. Lehmke, Arch. Pharm. (Weinheim, Ger.) 1985, 318, 11-14.
- ↑ 26.0 26.1 A. C. Campbell, M. S. Maidment, J. H. Pick, D. F. M. Stevenson, J. Chem. Soc., Perkin Trans. 1 1985, 1567-1576. doi:10.1039/P19850001567.
- ↑ 27.0 27.1 C. E. Caufield, S. A. Antane, K. M. Morris, S. M. Naughton, D. A. Quagliato, P. M. Andrae, A. Enos, J. F. Chiarello, J. (Wyeth, and Brother Ltd., USA), WO 2005019196, US 2005054721, 2005.
- ↑ 28.0 28.1 S. A. Antane, C. E. Caufield, W. Hu, D. Keeney, P. Labthavikul, K. M. Morris, S. M. Naughton, P. J. Petersen, B. A. Rasmussen, G. Singh, Y. Yang, Bioorg. Med. Chem. Lett. 2006, 176-180. doi:10.1016/j.bmcl.2005.09.021.
- ↑ 29.0 29.1 M. Gill, W. Steglich, Prog. Chem. Org. Nat. Prod., 1987, 51, 1-317. Springer-Verlag.
- ↑ D. W. Knight, G. Pattenden, J. Chem. Soc., Chem. Commun. 1975, 876-877 doi:10.1039/C39750000876.
- ↑ D. W. Knight, G. Pattenden, J. Chem. Soc., Perkin Trans. 1 1979, 70-76 doi:10.1039/P19790000070.
- ↑ P. J. Jerris, P. M. Wovkulich, A. B. Smith III, Tetrahedron Lett. 1979, 20, 4517-20 doi:10.1016/S0040-4039(01)86637-5.
- ↑ M. Gill, M. J. Kiefel, D. A. Lally, A. Ten, Aust. J. Chem. 1990, 43, 1497-518.
- ↑ G. Pattenden, M. W. Turvill, A. P. Chorlton, J. Chem. Soc., Perkin Trans. 1 1991, 10, 2357-2361. doi:10.1039/P19910002357.
- ↑ N. Kaczybura, R. Brückner Synthesis 2007, 118-130. doi:10.1055/s-2006-950378.
- ↑ D. Bernier, F. Moser, R. Brückner Synthesis 2007, 15, 2240-2248. doi:10.1055/s-2007-983800.
- ↑ D. Bernier, R. Brückner Synthesis 2007, 15, 2249-2272. doi:10.1055/s-2007-983803.
| ||||||
| ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||