Polycystic kidney disease
| Polycystic kidney disease | |
|---|---|
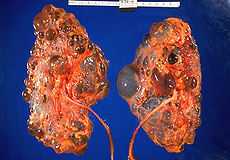 Polycystic kidneys | |
| Classification and external resources | |
| ICD-10 | Q61 |
| ICD-9 | 753.1 |
| OMIM | 173900 |
| DiseasesDB | 10262 10280 |
| MedlinePlus | 000502 |
| eMedicine | med/1862 ped/1846 radio/68 radio/69 |
| Patient UK | Polycystic kidney disease |
| MeSH | D007690 |
Polycystic kidney disease (PKD or PCKD, also known as polycystic kidney syndrome) is a cystic genetic disorder of the kidneys.[1] There are two types of PKD: autosomal dominant polycystic kidney disease (ADPKD) and the less-common autosomal recessive polycystic kidney disease (ARPKD).
It occurs in humans and some other animals. PKD is characterized by the presence of multiple cysts (hence, "polycystic") typically in both kidneys; however 17% of cases initially present with observable disease in one kidney, with most cases progressing to bilateral disease in adulthood.[2] The cysts are numerous and are fluid-filled, resulting in massive enlargement of the kidneys. The disease can also damage the liver, pancreas and, in some rare cases, the heart and brain. The two major forms of polycystic kidney disease are distinguished by their patterns of inheritance.
Polycystic kidney disease is one of the most common life-threatening genetic diseases, affecting an estimated 12.5 million people worldwide.[3]
Types
Polycystic Kidney Disease, or PKD, is a blanket term for the two types of PKD, each having their own pathology and causes. These two types of PKD are autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD).
Autosomal dominant

Autosomal dominant polycystic kidney disease (ADPKD) is the most common of all the hereditary cystic kidney diseases[2][4][5] with an incidence of 1:500 live births.[2][5] Studies show that 10% of end-stage renal disease (ESRD) patients being treated with hemodialysis in Europe and the U.S. were initially diagnosed and treated for ADPKD.[2]
In Brazil, the frequency of ADPKD is 10.3% among patients on dialysis. The Brazilian prevalence of ADPKD is low among hemodialysis patients, with 9.2 cases per 100,000 population (or 1: 10,912).[6] In regard to race, it was observed that about 70% of cases of ADPKD affect white people. There is no distinction between genders, that is, the distribution of cases of ADPKD is 50% among men and 50% among women.[7]
ADPKD is characterized by progressive cyst development and bilaterally enlarged kidneys with multiple cysts. There are three genetic mutations in the PKD-1, PKD-2, and PKD3 gene with similar phenotypical presentations. Gene PKD-1 is located on chromosome 16 and codes for a protein involved in regulation of cell cycle and intracellular calcium transport in epithelial cells, and is responsible for 85% of the cases of ADPKD. A group of voltage-linked calcium channels are coded for by PKD-2 on chromosome 4. PKD3 recently appeared in research papers as a postulated third gene. At this time, PKD3 has not been proven.[2][4] Fewer than 10% of cases of ADPKD appear in non-ADPKD families.
Cyst formation begins in utero from any point along the nephron, although fewer than 5% of nephrons are thought to be involved. As the cysts accumulate fluid, they enlarge, separate entirely from the nephron, compress the neighboring renal parenchyma, and progressively compromise renal function.
Under the function of gene defect, epithelial cells of renal tubule turn into epithelial cells of cyst wall after phenotype change, and begin to have the function of secreting cyst fluid, which leads to continuous cysts enlargement.[8]
Immunogenetics
The immunity is involved in the participation of the triggered response in ADPKD. The leukocytes contribute to local tissue damage in ADPKD due to the production of inflammatory mediators. Due to the chemotactic function of certain cytokines, the site of injury is characterized with leukocyte accumulation. The onset of kidney tissue injury occurs through the binding of antibodies to cell surface antigens, the matrix or basement membranes that dependent or independent of complement activation, initiates local inflammation at the early release cytokines. It has also been observed that the expression and production of cytokines are partially genetically determined. In recent years, allelic variants have been described for several genes of cytokines associated with the clinical progression of renal disease. In general, the data point to a possible influence on gene regulation and secretion of pro-inflammatory cytokines and anti that could modulate the risk of these diseases.
A recent study,[9] investigated the existence of a possible association between variants in genes of cytokines and cytokine receptors for various positions and ADPKD. There was influence on susceptibility or resistance to the predisposition of ADPKD the following variants in genes of proinflammatory cytokines: TNF-308, -238 (GG / GG [OR = 0.44]), -238 (G / G [OR = 0 35], G / A [OR = 2.84], G [OR = 0.38] and A [OR = 2.62]) and IL2-330, +166 (GG / GG [OR = 4.93 ]) -330 (G / G [OR = 2.56]) and anti-inflammatory TGFB1códon 10 (C / C [OR = 2.22] and C [OR = 1.66]) and IL4-1098 - 590, -33 (TCC / GCC [OR = 2.14]), -1098 (T / G [OR = 2.31]). The results show that cytokine SNPs in genes are determinant in the potential predisposition for ADPKD.
Autosomal recessive
Autosomal recessive polycystic kidney disease (ARPKD) (OMIM #263200) is the lesser common of the two types of PKD, with an incidence of 1:20,000 live births and is typically identified in the first few weeks after birth. Unfortunately, resulting hypoplasia results in a 30% death rate in neonates with ARPKD.[2] In ARPKD kidneys retain their shape but are larger than the normal anatomical range with dilated collecting ducts from the medulla to the cortex.
References
- ↑ "polycystic kidney disease" at Dorland's Medical Dictionary
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 Bisceglia, M et al. (2006). "Renal cystic diseases: a review". Advanced Anatomic Pathology (13): 26–56.
- ↑ Wilson PD. Polycystic kidney disease. N Engl J Med 2004;350:151-164
- ↑ 4.0 4.1 Torres, WE; Harris PC; Pirson Y (2007). "Autosomal dominant polycystic urology". Lancet 369 (9569): 1287–301. doi:10.1016/S0140-6736(07)60601-1.
- ↑ 5.0 5.1 Simons, M; Walz G (2006). "Polycystic kidney disease: cell division with a c(l)ue?". Kidney International 70: 854–864. doi:10.1038/sj.ki.5001534.
- ↑ Alves EF, Tsuneto LT, Pelloso SM, Torres PRA, Otto GLG, Silva AA, Obregon JMV, Silva LN, Carvalho MDB. Autosomal dominant polycystic kidney disease in hemodialysis patients in southern Brazil. J Bras Nefrol 2014; 36(1):18-25
- ↑ Alves EF, Tsuneto LT, Borelli SD, Cadidé RC, Freitas RA, Gravena AAF, Pelloso SM, Carvalho MDB. Características sociodemográficas e aspectos clínicos de pacientes com doença renal policística do adulto submetidos à hemodiálise. Sci Med. 2013;23(3):156-162
- ↑ 梅长林,常染色体显性多囊肾病,肾脏病学,第三版,人民卫生出版社|year=2008,9|pages=1746
- ↑ Alves EF. Estudo de associação entre os polimorfismos de um único nucleotídeo em genes de citocinas e a doença renal policística autossômica dominante em uma população brasileira. Dissertação (Mestrado em Ciências da Saúde). Maringá: UEM; 2014. 73p
External links
- Polycystic Kidney Disease Research and Education
- The National Kidney Foundation: A to Z Health Guide
- Enfermedad Polquística Renal en castellano
| ||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||