Phylogenetics
| Part of a series on |
| Evolutionary biology |
|---|
 Diagrammatic representation of the divergence of modern taxonomic groups from their common ancestor. |
|
Natural history
|
|
History of evolutionary theory |
|
Fields and applications
|
|
Social implications |
|
In biology, phylogenetics /faɪlɵdʒɪˈnɛtɪks/ is the study of evolutionary relationships among groups of organisms (e.g. species, populations), which are discovered through molecular sequencing data and morphological data matrices. The term phylogenetics derives from the Greek terms phylé (φυλή) and phylon (φῦλον), denoting "tribe", "clan", "race"[1] and the adjectival form, genetikós (γενετικός), of the word genesis (γένεσις) "origin", "source", "birth".
In fact, phylogenesis is the process, phylogeny is science on this process, and phylogenetics is phylogeny based on analysis of sequences of biological macromolecules (DNA, RNA and proteins, in the first).[2] The result of phylogenetic studies is a hypothesis about the evolutionary history of taxonomic groups: their phylogeny.[3]
Evolution is a process whereby populations are altered over time and may split into separate branches, hybridize together, or terminate by extinction. The evolutionary branching process may be depicted as a phylogenetic tree, and the place of each of the various organisms on the tree is based on a hypothesis about the sequence in which evolutionary branching events occurred. In historical linguistics, similar concepts are used with respect to relationships between languages; and in textual criticism with stemmatics.
Phylogenetic analyses have become essential to research on the evolutionary tree of life. For example, the RedToL aims at reconstructing the Red Algal Tree of Life. The National Science Foundation sponsors a project called the Assembling the Tree of Life (AToL) activity. The goal of this project is to determine evolutionary relationships across large groups of organisms throughout the history of life. The research on this project often involves large teams working across institutions and disciplines, and typically provides support to investigators working on computational phylogenetics and phyloinformatics tasks, including data acquisition, analysis, and algorithm development and dissemination.
Taxonomy—the classification, identification and naming of organisms—is usually richly informed by phylogenetics, but remains a methodologically and logically distinct discipline.[4] The degree to which taxonomies depend on phylogenies differs depending on the school of taxonomy: phenetics ignores phylogeny altogether, trying to represent the similarity between organisms instead; cladistics (phylogenetic systematics) tries to reproduce phylogeny in its classification without loss of information; evolutionary taxonomy tries to find a compromise between them in order to represent stages of evolution.
Construction of a phylogenetic tree
The scientific methods of phylogenetics are often grouped under the term cladistics. The most common ones are parsimony, maximum likelihood (ML), and MCMC-based Bayesian inference. All methods depend upon an implicit or explicit mathematical model describing the evolution of characters observed in the species included; all can be, and are, used for molecular data, wherein the characters are aligned nucleotide or amino acid sequences, and all but maximum likelihood (see below) can be, and are, used for phenotypic (morphological, chemical, and physiological) data (also called classical or traditional data).
Phenetics, popular in the mid-20th century but now largely obsolete, uses distance matrix-based methods to construct trees based on overall similarity in morphology or other observable traits (i.e. in the phenotype, not the DNA), which was often assumed to approximate phylogenetic relationships.
A comprehensive step-by-step protocol on constructing phylogenetic tree, including DNA/Amino Acid contiguous sequence assembly, multiple sequence alignment, model-test (testing best-fitting substitution models) and phylogeny reconstruction using Maximum Likelihood and Bayesian Inference, is available at Nature Protocol[5]
Prior to 1990, phylogenetic inferences were generally presented as narrative scenarios. Such methods are legitimate, but often ambiguous and hard to test.[6][7][8]
Limitations and workarounds
Ultimately, there is no way to measure whether a particular phylogenetic hypothesis is accurate or not, unless the true relationships among the taxa being examined are already known (which may happen with bacteria or viruses under laboratory conditions). The best result an empirical phylogeneticist can hope to attain is a tree with branches that are well supported by the available evidence. Several potential pitfalls have been identified:
Homoplasy
Certain characters are more likely to evolve convergently than others; logically, such characters should be given less weight in the reconstruction of a tree.[9] Weights in the form of a model of evolution can be inferred from sets of molecular data, so that maximum likelihood or Bayesian methods can be used to analyze them. For molecular sequences, this problem is exacerbated when the taxa under study have diverged substantially. As time since the divergence of two taxa increase, so does the probability of multiple substitutions on the same site, or back mutations, all of which result in homoplasies. For morphological data, unfortunately, the only objective way to determine convergence is by the construction of a tree – a somewhat circular method. Even so, weighting homoplasious characters does indeed lead to better-supported trees.[9] Further refinement can be brought by weighting changes in one direction higher than changes in another; for instance, the presence of thoracic wings almost guarantees placement among the pterygote insects because, although wings are often lost secondarily, there is no evidence that they have been gained more than once.[10]
Horizontal gene transfer
In general, organisms can inherit genes in two ways: vertical gene transfer and horizontal gene transfer. Vertical gene transfer is the passage of genes from parent to offspring, and horizontal (also called lateral) gene transfer occurs when genes jump between unrelated organisms, a common phenomenon especially in prokaryotes; a good example of this is the acquired antibiotic resistance as a result of gene exchange between various bacteria leading to multi-drug-resistant bacterial species. There have also been well-documented cases of horizontal gene transfer between eukaryotes.
Horizontal gene transfer has complicated the determination of phylogenies of organisms, and inconsistencies in phylogeny have been reported among specific groups of organisms depending on the genes used to construct evolutionary trees. The only way to determine which genes have been acquired vertically and which horizontally is to parsimoniously assume that the largest set of genes that have been inherited together have been inherited vertically; this requires analyzing a large number of genes.
Taxon sampling
Owing to the development of advanced sequencing techniques in molecular biology, it has become feasible to gather large amounts of data (DNA or amino acid sequences) to infer phylogenetic hypotheses. For example, it is not rare to find studies with character matrices based on whole mitochondrial genomes (~16,000 nucleotides, in many animals). However, simulations have shown that it is more important to increase the number of taxa in the matrix than to increase the number of characters, because the more taxa there are, the more accurate and more robust is the resulting phylogenetic tree.[11][12] This may be partly due to the breaking up of long branches.
Phylogenetic signal
Another important factor that affects the accuracy of tree reconstruction is whether the data analyzed actually contain a useful phylogenetic signal, a term that is used generally to denote whether a character evolves slowly enough to have the same state in closely related taxa as opposed to varying randomly. Tests for phylogenetic signal exist.[13]
Continuous characters
Morphological characters that sample a continuum may contain phylogenetic signal, but are hard to code as discrete characters. Several methods have been used, one of which is gap coding, and there are variations on gap coding.[14] In the original form of gap coding:[14]
group means for a character are first ordered by size. The pooled within-group standard deviation is calculated … and differences between adjacent means … are compared relative to this standard deviation. Any pair of adjacent means is considered different and given different integer scores … if the means are separated by a "gap" greater than the within-group standard deviation … times some arbitrary constant.
If more taxa are added to the analysis, the gaps between taxa may become so small that all information is lost. Generalized gap coding works around that problem by comparing individual pairs of taxa rather than considering one set that contains all of the taxa.[14]
Missing data
In general, the more data that are available when constructing a tree, the more accurate and reliable the resulting tree will be. Missing data are no more detrimental than simply having fewer data, although the impact is greatest when most of the missing data are in a small number of taxa. Concentrating the missing data across a small number of characters produces a more robust tree.[15]
The role of fossils
Because many characters involve embryological, or soft-tissue or molecular characters that (at best) hardly ever fossilize, and the interpretation of fossils is more ambiguous than that of living taxa, extinct taxa almost invariably have higher proportions of missing data than living ones. However, despite these limitations, the inclusion of fossils is invaluable, as they can provide information in sparse areas of trees, breaking up long branches and constraining intermediate character states; thus, fossil taxa contribute as much to tree resolution as modern taxa.[16] Fossils can also constrain the age of lineages and thus demonstrate how consistent a tree is with the stratigraphic record;[17] stratocladistics incorporates age information into data matrices for phylogenetic analyses.
History
The term "phylogeny" derives from the German Phylogenie, introduced by Haeckel in 1866.[18]
Ernst Haeckel's recapitulation theory
During the late 19th century, Ernst Haeckel's recapitulation theory, or "biogenetic fundamental law", was widely accepted. It was often expressed as "ontogeny recapitulates phylogeny", i.e. the development of an organism successively mirrors the adult stages of successive ancestors of the species to which it belongs. This theory has long been rejected.[19][20] Instead, ontogeny evolves – the phylogenetic history of a species cannot be read directly from its ontogeny, as Haeckel thought would be possible, but characters from ontogeny can be (and have been) used as data for phylogenetic analyses; the more closely related two species are, the more apomorphies their embryos share.
Timeline of key events

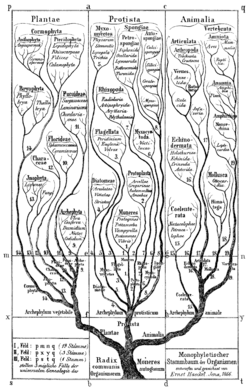
- 1300s, lex parsimoniae (parsimony principle), William of Ockam, English philosopher, theologian, and Franciscan monk, but the idea actually goes back to Aristotle, precursor concept
- 1763, Bayesian probability, Rev. Thomas Bayes,[21] precursor concept
- 1700s, Pierre Simon (Marquis de Laplace), perhaps 1st to use ML (maximum likelihood), precursor concept
- 1809, evolutionary theory, Philosophie Zoologique, Jean-Baptiste de Lamarck, precursor concept, foreshadowed in the 1600s and 1700s by Voltaire, Descartes, and Leibniz, with Leibniz even proposing evolutionary changes to account for observed gaps suggesting that many species had become extinct, others transformed, and different species that share common traits may have at one time been a single race,[22] also foreshadowed by some early Greek philosophers such as Anaximander in the 6th century BC and the atomists of the 5th century BC, who proposed rudimentary theories of evolution[23]
- 1837, Darwin's notebooks show an evolutionary tree[24]
- 1843, distinction between homology and analogy (the latter now referred to as homoplasy), Richard Owen, precursor concept
- 1858, Paleontologist Heinrich Georg Bronn (1800–1862) published a hypothetical tree to illustrating the paleontological "arrival" of new, similar species following the extinction of an older species. Bronn did not propose a mechanism responsible for such phenomena, precursor concept.[25]
- 1858, elaboration of evolutionary theory, Darwin and Wallace,[26] also in Origin of Species by Darwin the following year, precursor concept
- 1866, Ernst Haeckel, first publishes his phylogeny-based evolutionary tree, precursor concept
- 1893, Dollo's Law of Character State Irreversibility,[27] precursor concept
- 1912, ML recommended, analyzed, and popularized by Ronald Fisher, precursor concept
- 1921, Tillyard uses term "phylogenetic" and distinguishes between archaic and specialized characters in his classification system[28]
- 1940, term "clade" coined by Lucien Cuénot
- 1949, jackknife, Maurice Quenouille (foreshadowed in '46 by Mahalanobis and extended in '58 by Tukey), precursor concept
- 1950, Willi Hennig's classic formalization[29]
- 1952, William Wagner's groundplan divergence method[30]
- 1953, "cladogenesis" coined[31]
- 1960, "cladistic" coined by Cain and Harrison[32]
- 1963, 1st attempt to use ML (maximum likelihood) for phylogenetics, Edwards and Cavalli-Sforza[33]
- 1965
- 1966
- English translation of Hennig[36]
- "cladistics" and "cladogram" coined (Webster's, loc. cit.)
- 1969
- 1970, Wagner parsimony generalized by Farris[40]
- 1971
- Fitch parsimony, Fitch[41]
- NNI (nearest neighbour interchange), 1st branch-swapping search strategy, developed independently by Robinson[42] and Moore et al.
- ME (minimum evolution), Kidd and Sgaramella-Zonta[43] (it is unclear if this is the pairwise distance method or related to ML as Edwards and Cavalli-Sforza call ML "minimum evolution".)
- 1972, Adams consensus, Adams[44]
- 1974, 1st successful application of ML to phylogenetics (for nucleotide sequences), Neyman[45]
- 1976, prefix system for ranks, Farris[46]
- 1977, Dollo parsimony, Farris[47]
- 1979
- 1980, PHYLIP, 1st software package for phylogenetic analysis, Felsenstein
- 1981
- 1982
- PHYSIS, Mikevich and Farris
- branch and bound, Hendy and Penny[54]
- 1985
- 1986, MacClade, Maddison and Maddison
- 1987, neighbor-joining method Saitou and Nei[58]
- 1988, Hennig86 (version 1.5), Farris
- 1989
- 1990
- 1991
- 1993, implied weighting Goloboff[65]
- 1994, Bremer support (decay index), Bremer[66]
- 1994, reduced consensus: RCC (reduced cladistic consensus) for rooted trees, Wilkinson[67]
- 1995, reduced consensus RPC (reduced partition consensus) for unrooted trees, Wilkinson[68]
- 1996, 1st working methods for BI (Bayesian Inference)independently developed by Li,[69] Mau,[70] and Rannalla and Yang[71] and all using MCMC (Markov chain-Monte Carlo)
- 1998, TNT (Tree Analysis Using New Technology), Goloboff, Farris, and Nixon
- 1999, Winclada, Nixon
- 2003, symmetrical resampling, Goloboff[72]
See also
- Angiosperm Phylogeny Group
- Bauplan
- Bioinformatics
- Biomathematics
- Cladistics
- Coalescent theory
- Computational phylogenetics
- EDGE of Existence programme
- Evolutionary taxonomy
- Joe Felsenstein
- Language family
- Maximum parsimony
- Microbial phylogenetics
- Molecular phylogeny
- Ontogeny
- Ontogeny (psychoanalysis)
- PhyloCode
- Phylodynamics
- Phylogenetic comparative methods
- Phylogenetic network
- Phylogenetic nomenclature
- Phylogenetic tree
- Phylogenetic tree viewers
- Phylogenetics software
- Phylogenomics
- Phylogeny (psychoanalysis)
- Phylogeography
- Systematics
References
- ↑ Liddell, Henry George; Scott, Robert; Jones, Henry Stuart (1968). A Greek-English lexicon (9 ed.). Oxford: Clarendon Press. p. 1961.
- ↑ Liddell, Henry George; Scott, Robert; Jones, Henry Stuart (1968). A Greek-English lexicon (9 ed.). Oxford: Clarendon Press. p. 343.
- ↑ "phylogeny". Biology online. Retrieved 2013-02-15.
- ↑ Edwards AWF; Cavalli-Sforza LL (1964). "Reconstruction of evolutionary trees". In Heywood, Vernon Hilton; McNeill, J. Phenetic and Phylogenetic Classification. pp. 67–76. OCLC 733025912.
Phylogenetics is the branch of life science concerned with the analysis of molecular sequencing data to study evolutionary relationships among groups of organisms.
- ↑ Bast, F. 2013. Sequence Similarity Search, Multiple Sequence Alignment, Model Selection, Distance Matrix and Phylogeny Reconstruction. Nature Protocol Exchange. doi:10.1038/protex.2013.065
- ↑ Richard C. Brusca & Gary J. Brusca (2003). Invertebrates (2nd ed.). Sunderland, Massachusetts: Sinauer Associates. ISBN 978-0-87893-097-5.
- ↑ Bock, W.J. (2004). Explanations in systematics. Pp. 49-56. In Williams, D.M. and Forey, P.L. (eds) Milestones in Systematics. London: Systematics Association Special Volume Series 67. CRC Press, Boca Raton, Florida.
- ↑ Auyang, Sunny Y. (1998). Narratives and Theories in Natural History. In: Foundations of complex-system theories: in economics, evolutionary biology, and statistical physics. Cambridge, U.K.; New York: Cambridge University Press.
- ↑ 9.0 9.1 Goloboff, Pablo A.; Carpenter, James M.; Arias, J. Salvador; Esquivel, Daniel Rafael Miranda (2008). "Weighting against homoplasy improves phylogenetic analysis of morphological data sets". Cladistics 24 (5): 758. doi:10.1111/j.1096-0031.2008.00209.x.
- ↑ Goloboff, Pablo A. (1997). "Self-Weighted Optimization: Tree Searches and Character State Reconstructions under Implied Transformation Costs". Cladistics 13 (3): 225. doi:10.1111/j.1096-0031.1997.tb00317.x.
- ↑ Zwickl, Derrick J.; Hillis, David M. (2002). "Increased Taxon Sampling Greatly Reduces Phylogenetic Error". Systematic Biology 51 (4): 588–98. doi:10.1080/10635150290102339. PMID 12228001.
- ↑ Wiens, John J. (2006). "Missing data and the design of phylogenetic analyses". Journal of Biomedical Informatics 39 (1): 34–42. doi:10.1016/j.jbi.2005.04.001. PMID 15922672.
- ↑ Blomberg, Simon P.; Garland Jr, Theodore; Ives, Anthony R. (2003). "Testing for phylogenetic signal in comparative data: Behavioral traits are more labile". Evolution 57 (4): 717–45. doi:10.1111/j.0014-3820.2003.tb00285.x. PMID 12778543.
- ↑ 14.0 14.1 14.2 Archie, J.W. (1985). "Methods for coding variable morphological features for numerical taxonomic analysis". Systematic Zoology 34 (3): 326–345. doi:10.2307/2413151.
- ↑ Prevosti, Francisco J.; Chemisquy, María A. (2009). "The impact of missing data on real morphological phylogenies: Influence of the number and distribution of missing entries". Cladistics 26 (3): 326. doi:10.1111/j.1096-0031.2009.00289.x.
- ↑ Cobbett, Andrea; Wilkinson, Mark; Wills, Matthew (2007). "Fossils Impact as Hard as Living Taxa in Parsimony Analyses of Morphology". Systematic Biology 56 (5): 753–66. doi:10.1080/10635150701627296. PMID 17886145.
- ↑ Huelsenbeck, John P. (1994). "Comparing the Stratigraphic Record to Estimates of Phylogeny". Paleobiology 20 (4): 470–83. JSTOR 2401230.
- ↑ Harper, Douglas (2010). "Phylogeny". Online Etymology Dictionary. Retrieved March 18, 2013.
- ↑ Blechschmidt, Erich (1977) The Beginnings of Human Life. Springer-Verlag Inc., p. 32: "The so-called basic law of biogenetics is wrong. No buts or ifs can mitigate this fact. It is not even a tiny bit correct or correct in a different form, making it valid in a certain percentage. It is totally wrong."
- ↑ Ehrlich, Paul; Richard Holm; Dennis Parnell (1963) The Process of Evolution. New York: McGraw–Hill, p. 66: "Its shortcomings have been almost universally pointed out by modern authors, but the idea still has a prominent place in biological mythology. The resemblance of early vertebrate embryos is readily explained without resort to mysterious forces compelling each individual to reclimb its phylogenetic tree."
- ↑ Bayes, T. 1763. An Essay towards solving a Problem in the Doctrine of Chances. Phil. Trans. 53: 370–418.
- ↑ Strickberger, Monroe. 1996. Evolution, 2nd. ed. Jones & Bartlett.
- ↑ The Theory of Evolution, Teaching Company course, Lecture 1
- ↑ Darwin's Tree of Life
- ↑ J. David Archibald (2009) 'Edward Hitchcock’s Pre-Darwinian (1840) 'Tree of Life'.', Journal of the History of Biology (2009) page 568.
- ↑ Darwin, C. R. and A. R. Wallace. 1858. On the tendency of species to form varieties; and on the perpetuation of varieties and species by natural means of selection. Journal of the Proceedings of the Linnean Society of London. Zoology 3: 45-50.
- ↑ Dollo, Louis. 1893. Les lois de l'évolution. Bull. Soc. Belge Géol. Paléont. Hydrol. 7: 164-66.
- ↑ Tillyard R. J. 1921. A new classification of the order Perlaria. Canadian Entomologist 53: 35-43
- ↑ Hennig. W. (1950). Grundzuge einer theorie der phylogenetischen systematik. Deutscher Zentralverlag, Berlin.
- ↑ Wagner, W.H. Jr. 1952. The fern genus Diellia: structure, affinities, and taxonomy. Univ. Calif. Publ. Botany 26: 1–212.
- ↑ Webster's 9th New Collegiate Dictionary
- ↑ Cain, A. J., Harrison, G. A. 1960. "Phyletic weighting". Proceedings of the Zoological Society of London 35: 1–31.
- ↑ Edwards, A.W.F, Cavalli-Sforza, L.L. (1963). The reconstruction of evolution. Ann. Hum. Genet. 27: 105–106.
- ↑ Camin J.H, Sokal R.R. (1965). A method for deducing branching sequences in phylogeny.Evolution 19: 311–326.
- ↑ Wilson, E. O. 1965. A consistency test for phylogenies based on contemporaneous species. Systematic Zoology 14: 214-220.
- ↑ Hennig. W. (1966). Phylogenetic systematics. Illinois University Press, Urbana.
- ↑ Farris, J.S. 1969. A successive approximations approach to character weighting. Syst. Zool. 18: 374-85.
- ↑ 38.0 38.1 Kluge, A.G, Farris, J.S. (1969). Quantitative phyletics and the evolution of anurans. Syst. Zool. 18: 1–32.
- ↑ Le Quesne, W. J. 1969. A method of selection of characters in numerical taxonomy. Systematic Zoology 18: 201-205.
- ↑ Farris, J.S. (1970). Methods of computing Wagner trees. Syst. Zool. 19: 83–92.
- ↑ Fitch, W.M. (1971). Toward defining the course of evolution: minimum change for a specified tree topology. Syst. Zool. 20: 406–416.
- ↑ Robinson. D.F. 1971. Comparison of labeled trees with valency three. Journal of Combinatorial Theory 11:105–119.
- ↑ Kidd, K.K. and Laura Sgaramella-Zonta (1971). Phylogenetic analysis: concepts and methods. Am. J. Human Genet. 23, 235-252.
- ↑ Adams, E. (1972). Consensus techniques and the comparison of taxonomic trees. Syst. Zool. 21: 390–397.
- ↑ Neyman, J. (1974). Molecular studies: A source of novel statistical problems. In: Gupta SS, Yackel J. (eds), Statistical Decision Theory and Related Topics, pp. 1–27. Academic Press, New York.
- ↑ Farris, J.S. (1976). Phylogenetic classification of fossils with recent species. Syst. Zool. 25: 271-282.
- ↑ Farris, J.S. (1977). Phylogenetic analysis under Dollo’s Law. Syst. Zool. 26: 77–88.
- ↑ Nelson, G.J. 1979. Cladsitic analysis and synthesis: pronciples and definitions with a historical noteon Adanson's Famille des plantes (1763-1764). Syst. Zool. 28: 1-21.
- ↑ Gordon, Aé<.D. 1979. A measure of the agreement between rankings. Biometrika 66: 7-15.
- ↑ Efron B. (1979). Bootstrap methods: another look at the jackknife. Ann. Stat. 7: 1–26.
- ↑ Margush T, McMorris FR. 1981. Consensus n-trees. Bull. Math .Biol. 43, 239–244.
- ↑ Sokal, R. R., F. J. Rohlf. 1981. Taxonomic congruence in the Leptopodomorpha re-examined. Syst. Zool. 30:309-325.
- ↑ Felsenstein, J. (1981). Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 17: 368–376.
- ↑ Hendy MD, Penny D (1982) Branch and bound algorithms to determine minimal evolutionary trees. Math Biosci 59: 277–290.
- ↑ Lipscomb, Diana. 1985. The Eukaryotic Kingdoms. Cladistics 1: 127-40.
- ↑ Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791.
- ↑ Lanyon, S.M. (1985). Detecting internal inconsistencies in distance data. Syst. Zool. 34: 397-403.
- ↑ Saitou N, Nei M (1987) The Neighbor-joining Method: A New Method for Constructing Phylogenetic Trees. Mol. Biol. Evol. 4:406-425.
- ↑ Farris, J.S. (1989). The retention index and rescaled consistency index. Cladistics 5: 417–419.
- ↑ Archie, J.W. 1989. Homoplasy Excess Ratios: new indices for measuring levels of homoplasy in phylogentic systematics and a critique of the Consistency Index. Syst. Zool. 38: 253-69.
- ↑ Bremer. Kåre. 1990. Combinable Component Consensus. Cladistics 6: 369–372.
- ↑ D.L. Swofford and G.J. Olsen. 1990. Phylogeny reconstruction. In D.M. Hillis andG. Moritz, editors, Molecular Systematics, pages 411–501. Sinauer Associates, Sunderland, Mass.
- ↑ Goloboff, P. A. (1991). Homoplasy and the choice among cladograms. Cladistics 7:215–232.
- ↑ Goloboff, P. A. (1991b). Random data, homoplasy and information.Cladistics 7:395–406.
- ↑ Goloboff, P. A. 1993. Estimating character weights during tree search. Cladistics 9: 83–91.
- ↑ Bremer, K. 1994. Branch support and tree stability.
- ↑ Wilkinson, Mark. 1994. Common cladistic information and its consensus representation: reduced Adams and reduced cladistic consensus trees and profiles. Syst. Biol. 43:343-368.
- ↑ Wilkinson, Mark. 1995. More on reduced consensus methods. Syst. Biol. 44:436-440.
- ↑ Li, S. (1996). Phylogenetic tree construction using Markov Chain Monte Carlo. Ph.D. disseration, Ohio State University, Columbus.
- ↑ Mau B (1996) Bayesian phylogenetic inference via Markov chain Monte Carlo Methods. Ph.D. dissertation, University of Wisconsin, Madison (abstract).
- ↑ Rannala B, Yang Z. 1996. Probability distribution of molecular evolutionary trees: A new method of phylogenetic inference. J. Mol. Evol. 43: 304–311.
- ↑ Goloboff, Pablo; Farris, James; Källersjö, Mari; Oxelman, Bengt; Ramiacuterez, Maria; Szumik, Claudia. 2003. Improvements to resampling measures of group support. Cladistics 19: 324–332.
Further reading
- Schuh, Randall T.; Brower, Andrew V.Z. (2009). Biological Systematics: principles and applications (2nd ed.). Ithaca: Comstock Pub. Associates/Cornell University Press. ISBN 978-0-8014-4799-0. OCLC 312728177.
- Forster, Peter; Renfrew, Colin, eds. (2006). Phylogenetic Methods and the Prehistory of Languages. McDonald Institute Press, University of Cambridge. ISBN 978-1-902937-33-5. OCLC 69733654.
- Baum, David A.; Smith, Stacey D. (2013). Tree Thinking: an introduction to phylogenetic biology. Greenwood Village, CO: Roberts and Company. ISBN 978-1-936221-16-5. OCLC 767565978.
External links
| Look up phylogenetics in Wiktionary, the free dictionary. |
- The Tree of Life
- Interactive Tree of Life
- PhyloCode
- ExploreTree
- UCMP Exhibit Halls: Phylogeny Wing
- Willi Hennig Society
- Filogenetica.org in Spanish
- PhyloPat, Phylogenetic Patterns
- SplitsTree, program for computing phylogenetic trees and unrooted phylogenetic networks
- Dendroscope, program for drawing phylogenetic trees and rooted phylogenetic networks
- Phylogenetic inferring on the T-REX server
- Mesquite
- NCBI – Systematics and Molecular Phylogenetics
- Sydney Brenner's seminars: "What Genomes Can Tell Us About the Past"
- Mikko's Phylogeny Archive
- Who is Who in Phylogenetic Networks research papers related to the phylogenetic network
- Phylogenetic Reconstruction from Gene-Order Data
- ETE: A Python Environment for Tree Exploration This is a programming library to analyze, manipulate and visualize phylogenetic trees. See: Huerta-Cepas, Jaime; Dopazo, Joaquín; Gabaldón, Toni (2010). "ETE: A python Environment for Tree Exploration". BMC Bioinformatics 11: 24. doi:10.1186/1471-2105-11-24. PMC 2820433. PMID 20070885.
- PhylomeDB: A public database hosting thousands of gene phylogenies ranging many different species. See: Huerta-Cepas, J.; Capella-Gutierrez, S.; Pryszcz, L. P.; Denisov, I.; Kormes, D.; Marcet-Houben, M.; Gabaldón, T. (2010). "PhylomeDB v3.0: An expanding repository of genome-wide collections of trees, alignments and phylogeny-based orthology and paralogy predictions". Nucleic Acids Research 39 (Database issue): D556–60. doi:10.1093/nar/gkq1109. PMC 3013701. PMID 21075798.
- Lents, N. H.; Cifuentes, O. E.; Carpi, A. (2010). "Teaching the Process of Molecular Phylogeny and Systematics: A Multi-Part Inquiry-Based Exercise". Cell Biology Education 9 (4): 513. doi:10.1187/cbe.09-10-0076.
| ||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||