Periodic boundary conditions

Periodic boundary conditions (PBCs) are a set of boundary conditions which are often chosen for approximating a large (infinite) system by using a small part called a unit cell. PBCs are often used in computer simulations and mathematical models. The topology of two-dimensional PBC is equal to that of a world map of some video games; the geometry of the unit cell satisfies perfect two-dimensional tiling, and when an object passes through one side of the unit cell, it re-appears on the opposite side with the same velocity. In topological terms, the space made by two-dimensional PBCs can be thought of as being mapped onto a torus (compactification). The large systems approximated by PBCs consist of an infinite number of unit cells. In computer simulations, one of these is the original simulation box, and others are copies called images. During the simulation, only the properties of the original simulation box need to be recorded and propagated. The minimum-image convention is a common form of PBC particle bookkeeping in which each individual particle in the simulation interacts with the closest image of the remaining particles in the system.
One example of periodic boundary conditions can be defined according to smooth real functions  by
by


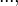

for all m = 0, 1, 2, ... and for constants 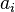 and
and 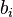 .
.
In molecular dynamics simulation, PBC are usually applied to calculate bulk gasses, liquids, crystals or mixtures. A common application uses PBC to simulate solvated macromolecules in a bath of explicit solvent. Born-von Karman boundary conditions are periodic boundary conditions for a special system.
PBC requirements and artifacts
Three-dimensional PBCs are useful for approximating the behavior of macro-scale systems of gases, liquids, and solids. Three-dimensional PBCs can also be used to simulate planar surfaces, in which case two-dimensional PBCs are often more suitable. Two-dimensional PBCs for planar surfaces are also called slab boundary conditions; in this case, PBCs are used for two Cartesian coordinates (e.g., x and y), and the third coordinate (z) extends to infinity.
PBCs can be used in conjunction with Ewald summation methods (e.g., the particle mesh Ewald method) to calculate electrostatic forces in the system. However, PBCs also introduce correlational artifacts that do not respect the translational invariance of the system,[1] and requires constraints on the composition and size of the simulation box.
In simulations of solid systems, the strain field arising from any inhomogeneity in the system will be artificially truncated and modified by the periodic boundary. Similarly, the wavelength of sound or shock waves and phonons in the system is limited by the box size.
In simulations containing ionic (Coulomb) interactions, the net electrostatic charge of the system must be zero to avoid summing to an infinite charge when PBCs are applied. In some applications it is appropriate to obtain neutrality by adding ions such as sodium or chloride (as counterions) in appropriate numbers if the molecules of interest are charged. Sometimes ions are even added to a system in which the molecules of interest are neutral, to approximate the ionic strength of the solution in which the molecules naturally appear. Maintenance of the minimum-image convention also generally requires that a spherical cutoff radius for nonbonded forces be at most half the length of one side of a cubic box. Even in electrostatically neutral systems, a net dipole moment of the unit cell can introduce a spurious bulk-surface energy, equivalent to pyroelectricity in polar crystals.
The size of the simulation box must also be large enough to prevent periodic artifacts from occurring due to the unphysical topology of the simulation. In a box that is too small, a macromolecule may interact with its own image in a neighboring box, which is functionally equivalent to a molecule's "head" interacting with its own "tail". This produces highly unphysical dynamics in most macromolecules, although the magnitude of the consequences and thus the appropriate box size relative to the size of the macromolecules depends on the intended length of the simulation, the desired accuracy, and the anticipated dynamics. For example, simulations of protein folding that begin from the native state may undergo smaller fluctuations, and therefore may not require as large a box, as simulations that begin from a random coil conformation. However, the effects of solvation shells on the observed dynamics – in simulation or in experiment – are not well understood. A common recommendation based on simulations of DNA is to require at least 1 nm of solvent around the molecules of interest in every dimension.[2]
Practical implementation: continuity and the minimum image convention
To implement a PBC algorithm, at least two steps are needed.
First, an object which has passed through one face of the simulation box should re-enter the box through the opposite face. This is a simple operation which can be described with the following code, where x_size is the length of the box in one direction (assuming an orthogonal unit cell centered on the origin) and x is the position of the particle in the same direction:
if (periodic_x) then if (x < -x_size * 0.5) x = x + x_size if (x >= x_size * 0.5) x = x - x_size end if
Second, every distance between objects should obey the minimum image criterion. This can be implemented according to the following code (in the case of a one-dimensional system where dx is the distance between object i and object j):
if (periodic_x) then dx = x(j) - x(i) if (abs(dx) > x_size * 0.5) dx = dx - sign(x_size, dx) end if
For three-dimensional PBCs, both operations should be repeated in all 3 dimensions.
These operations can be written in a much more compact form for orthorhombic cells if the origin is shifted to a corner of the box. Then we have, in one dimension, for positions and distances respectively:
! After x(i) update without regard to PBC: x(i) = x(i) - floor(x(i) / x_size) * x_size ! For a box with the origin at the lower left vertex ! Works for x's lying in any image. dx = x(j) - x(i) dx = dx - nint(dx / x_size) * x_size
For non-orthorhombic cells the situation is more complicated.[3]
In simulations of ionic systems more complicated operations may be needed to handle the long-range Coulomb interactions spanning several box images.
Unit cell geometries
PBC requires the unit cell to be a shape that will tile perfectly into a three-dimensional crystal. Thus, a spherical or elliptical droplet cannot be used. A cube or rectangular prism is the most intuitive and common choice, but can be computationally expensive due to unnecessary amounts of solvent molecules in the corners, distant from the central macromolecules. A common alternative that requires less volume is the truncated octahedron.
Conserved properties
Under periodic boundary conditions, the linear momentum of the system is conserved. Angular momentum is not conserved. Conventional explanation of this fact is based on Noether's theorem. The theorem states that conservation of angular momentum follows from rotational invariance of Lagrangian. However in a paper [4] it is shown that this approach is not consistent. It fails to explain the absence of conservation of angular momentum of a single particle moving in a periodic cell. Lagrangian of the particle is constant and therefore rotationally invariant, while angular momentum of the particle is not conserved. This contradiction is caused by the fact that Noether's theorem is usually formulated for closed systems. The periodic cell exchanges mass momentum, angular momentum, and energy with the neighboring cells.
When applied to the microcanonical ensemble (constant particle number, volume, and energy, abbreviated NVE), using PBC rather than reflecting walls slightly alters the sampling of the simulation due to the conservation of total linear momentum and the position of the center of mass; this ensemble has been termed the "molecular dynamics ensemble"[5] or the NVEPG ensemble.[6] These additional conserved quantities introduce minor artifacts related to the statistical mechanical definition of temperature, the departure of the velocity distributions from a Boltzmann distribution, and violations of equipartition for systems containing particles with heterogeneous masses. The simplest of these effects is that a system of N particles will behave, in the molecular dynamics ensemble, as a system of N-1 particles. These artifacts have quantifiable consequences for small toy systems containing only perfectly hard particles; they have not been studied in depth for standard biomolecular simulations, but given the size of such systems, the effects will be largely negligible.[6]
Notes
- ↑ Cheatham TE, Miller JH, Fox T, Darden PA, Kollman PA. (1995). Molecular Dynamics Simulations on Solvated Biomolecular Systems: The Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, RNA, and Proteins. J Am Chem Soc 117:4193.
- ↑ de Souza ON, Ornstein RL. (1997). Effect of periodic box size on aqueous molecular dynamics simulation of a DNA dodecamer with particle-mesh Ewald method. Biophys J 72(6):2395-7. PMID 9168016
- ↑ Minimum image convention in non-cubic simulation cells
- ↑ Kuzkin V.A. (2014). On angular momentum balance in particle systems with periodic boundary conditions, ZAMM, 2014, DOI: 10.1002/zamm.201400045.
- ↑ Erpenbeck JJ, Wood WW. (1977). Statistical Mechanics, Part B: Time-dependent Processes, Modern Theoretical Chemistry Vol 6. ed. Berne BJ. Plenum, New York, USA. See pp1-40.
- ↑ 6.0 6.1 Shirts RB, Burt SR, Johnson AM. (2006). Periodic boundary condition induced breakdown of the equipartition principle and other kinetic effects of finite sample size in classical hard-sphere molecular dynamics simulation. J Chem Phys 125(16):164102. PMID 17092058
See also
- Helical boundary conditions
- Molecular modeling
- Software for molecular mechanics modeling
References
- Schlick T. (2002). Molecular Modeling and Simulation: An Interdisciplinary Guide. Interdisciplinary Applied Mathematics series, vol. 21. Springer: New York, NY, USA. ISBN 0-387-95404-X. See esp. pp272–6.
- Rapaport DC. (2004). The Art of Molecular Dynamics Simulation. 2nd ed. Cambridge University Press. ISBN 0-521-82568-7. See esp. pp15–20.
- Kuzkin V.A. (2014). On angular momentum balance in particle systems with periodic boundary conditions, ZAMM, 2014, DOI: 10.1002/zamm.201400045.