Neuroacanthocytosis
| Neuroacanthocytosis | |
|---|---|
| Classification and external resources | |
| OMIM | 200150 |
| DiseasesDB | 29707 |
| eMedicine | article/1150817 |
| MeSH | D054546 |
Neuroacanthocytosis is a group of genetically diverse conditions complicated by movement disorders, neurological problems and spiculated (misshapen) red blood cells. These syndromes, which include chorea acanthocytosis, McLeod syndrome, Huntington’s disease–like 2 (HDL2), and pantothenate kinase-associated neurodegeneration (PKAN), primarily affect the brain and the basal ganglia. The conditions are caused by genetic mutations of several different genes including, VPS13A, XK, JPH3 and PANK2. The mutations are inherited through various genetic mechanisms.
Specific neurologic symptoms characterize these diseases. These symptoms may include: involuntary or slow movement; posture and skeletal related abnormalities; weakness; cognitive impairment; psychiatric symptoms; and other symptoms related to brain degeneration and movement difficulties. The disorders all have in common the presence of spiculated red blood cells, also known as spur cells, which are formally called acanthocytes.
The diseases are hereditary, but rare, and in some cases extremely rare, with insufficient data to draw conclusions about frequency of the mutation. Huntington's disease-like 2 has slightly higher ethnic prevalence in South Africans and there is gender prevalence in McLeod Syndrome with males being more susceptible to the disease than females. The other two do not show any ethnic or gender bias.
Classification
| Neuroacanthocytosis disease group | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Disease | Mutation | Inheritance | ||||||||
| Chorea acanthocytosis | VPS13A (CHAC gene) | autosomal recessive | ||||||||
| McLeod syndrome | XK gene on X-chromosome | X-linked recessive | ||||||||
| Huntington’s disease–like 2 | JPH3 | autosomal dominant | ||||||||
| Pantothenate kinase-associated neurodegeneration | PANK2 | autosomal recessive | ||||||||
Four syndromes are classified as neuroacanthocytosis. These syndromes are caused by different genetic mutations, but the signs and symptoms are usually similar, leading to the unified classification as forms of neuroacanthocytosis.[1]
Signs and symptoms
Signs and symptoms of a neuroacanthocytosis diseases may vary slightly from case to case but usually include several predominant symptoms. The hallmark feature of a neuroacanthocytosis disease is the presence of Acanthocytes. Acanthocytosis originated from the Greek word acantha, meaning thorn. Acanthocytes are spiculated red blood cells and can be caused by altered distribution of membrane lipids or membrane protein/skeleton abnormalities. In neuroacanthocytosis, acanthocytes are caused by skeleton abnormalities in the membrane of the cells that are affected, causing them to take on a spiculated shape while the lipids in the cell membrane have no abnormalities.[2] Chorea, involuntary dance-like movement, is another very common symptom of neuroacanthocytosis. Affected individuals may also suffer from involuntary face and tongue movements, which can cause difficulties with speech and eating. These movements are usually abrupt and irregular and present during both rest and sleep.[3]
Individuals with neuroacanthocytosis also usually suffer from parkinsonism, the uncontrolled slowness of movements, and dystonia, abnormal body postures. Patients may have difficulty walking due to muscle weakness and the involuntary and uncontrollable movement complications caused by parkinsonism and chorea. Most affected individuals also have cognitive (intellectual) impairment and psychiatric symptoms such as anxiety, paranoia, depression, obsessive behavior, and pronounced emotional instability.[4] Seizures may also be a symptom of neuroacanthocytosis.[1]
Onset
The onset of a neuroacanthocytosis disease is usually between ages 20 and 40 with an average onset of symptoms occurring at age 32.[5] However, symptoms may occur as early as age ten in some individuals with atypical versions of the disease.[3] Affected individuals usually live for 10–20 years after onset occurs but complications from other illnesses can reduce their lifespan even more.[6]
Management
Currently, no treatment slows the neurodegeneration in neuroacanthocytosis disorders. Medication may be administered to decrease the involuntary movements produced by these syndromes. Antipsychotics are used to block dopamine, anticonvulsants treat seizures and botulinum toxin injections may control dystonia. Patients usually receive speech, occupational and physical therapies to help with the complications associated with movement. Sometimes, physicians will prescribe antidepressants for the psychological problems that accompany neuroacanthocytosis.[1] Recently, in December 2012, German neurosurgeons implanted electrodes in the bilateral pallidum in one neuroacanthocytosis patient to apply deep brain stimulation for counteracting the dyskinesic symptoms.[7] This greatly reduced the patient's involuntary movements, enabling him to walk, speak, and feed himself again, i.e., to live on his own again, which had been impossible for him before surgery. Though deep brain stimulation always brings the risk of infection with it,[8] it might be considered for alternative treatment.
Prognosis
Neuroacanthocytosis is a progressive disease in which patients usually develop significant cardiac or neurologic complications.[6] They also become prone to such ancillary illnesses as pneumonia that aggravate symptoms. Death usually occurs within five to ten years after the onset of severe symptoms. Cardiac and neurologic complications are the usual cause of death, but if patients do not develop such problems, their lifespan may be longer.[6] The disease may worsen with poor nutrition and complications of movement disorders or associated psychological disorders.[1]
Chorea acanthocytosis
Chorea acanthocytosis is a disease that affects movement in many parts of the body. The symptoms are mostly consistent with the symptoms prevalent in neuroacanthocytosis disorders, also, many people with chorea acanthocytosis uncontrollably bite their tongue, lips, and the inside of the mouth. Behaviorial changes are an early indicator of chorea acanthocytosis.[9]
Characteristics
Chorea acanthocytosis is diagnosed in individuals with the following manifestations:
- Progressive dystonia
- Tongue protrusion and tongue and lip biting
- Progressive cognitive and behavioral changes
- Progressive myopathy (muscle damage) characterized by muscle wasting (inability to properly use muscles) and weakness
- Eye movement abnormalities
- Acanthocytosis
- Since chorea acanthocytosis is an autosomal recessive disorder, positive family history for chorea acanthocytosis will confirm the diagnosis.[10]
Genetics
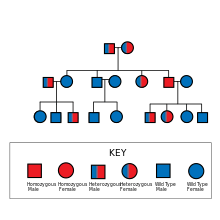
The mutation that causes chorea acanthocytosis is passed in an autosomal recessive manner, meaning that both alleles of the gene must be mutated for the person to have the disease. If both parents are affected by chorea acanthocytosis, all of the children will also acquire the disease. However, both parents may remain unaffected and be carriers of the mutated gene (heterozygous for the trait, carrying one defective allele) and produce an affected child. The likelihood of this occurring is 25%.[9]
The gene that determines whether or not a person has chorea acanthocytosis is known as VPS13A (CHAC gene) and is located on chromosome 9q21.[11] When mutated, this gene causes chorea acanthocytosis in the affected individual. This mutation causes a small, nonfunctional form of the protein chorein to be produced. Many researchers believe that chorein is responsible for cell movement, but the actual function of this protein remains unknown.[9]
Management
- Botulinum toxin injections (Botox) to reduce dystonia
- Mechanical protective devices (such as mouth guards) for tongue and lips
- Anticonvulsant drugs for seizure control
- Antipsychotics are prescribed for psychiatric problems
- Dopamine antagonists are prescribed (but should be heavily monitored) to patients to suppress involuntary movement[10]
Epidemiology
The appearance of symptoms in chorea acanthocytosis usually appear in early to mid-adulthood. The first sign of chorea acanthocytosis is often behavioral changes that result in personality changes, obsessive compulsive disorder (OCD), and the inability to take care of oneself. These continue on throughout the individual's life and movement complications beginning in early-adulthood worsen with age.[9] There are about 500–1,000 cases of chorea acanthocytosis worldwide and it is not specific to any particular ethnic group.[9]
Pantothenate kinase-associated neurodegeneration
Pantothenate kinase-associated neurodegeneration (PKAN) is a disease characterized by brain iron accumulation and progressive difficulty with movement. The symptoms of this disease are mostly consistent with the general symptoms of neuroacanthocytosis disorders. The affected individual may develop dementia and experience problems with vision loss. There is a classical version of PKAN and an atypical version in which onset occurs much later and is less severe. Pantothenate kinase-associated neurodegeneration was formerly known as Hallervorden-Spatz syndrome after two German neuropathologists but the term is no longer used because of the unethical activities these men participated in both before and during World War II.[12]
Characteristics
Individuals are diagnosed with Pantothenate kinase-associated neurodegeneration when they have the following manifestations:
- Dystonia and rigidity of movements
- Loss of ambulation (occurring within 15 years after onset)
- Eye of the tiger’s sign seen on an MRI caused by excess iron deposition in the globus pallidus (spherical section of the brain)[5]
- Retinal degeneration
- Corticospinal tract damage
- Acanthocytosis
- Family history of PKAN[13]
Genetics
Pantothenate kinase-associated neurodegeneration is inherited in an autosomal recessive pattern meaning that both copies of the gene in an individual must be mutated for the person to be affected by the disease. This means that if both parents are affected, all of the children will acquire the disease as well. However, if both parents are carriers of the disease (heterozygous for the trait, have one mutated copy) and are not affected by PKAN, there is a 25% chance that their child will acquire the disease.[12]
The gene responsible for Pantothenate kinase-associated neurodegeneration is known as PANK2. This gene aids in producing the enzyme pantothenate kinase 2 which is active in the mitochondria of cells. When active, pantothenate kinase 2 codes for coenzyme A which is essential for production of energy inside the organism from carbohydrates, fats and amino acids. When PANK2 is mutated, pantothenate kinase 2 does not function properly and therefore does not produce a working version of coenzyme A, allowing dangerous materials (like iron) to build up inside the brain instead of being converted into energy. This causes the symptoms that are present in Pantothenate kinase-associated neurodegeneration to occur.[12]
Prevention
There are no known prevention methods for Pantothenate kinase-associated neurodegeneration but certain drugs such as alpha-tocopherol and idebenone were shown to have worsened the symptoms of PKAN and should be avoided.[13]
Management
Most of the treatment for Pantothenate kinase-associated neurodegeneration is aimed at suppressing dystonia. Measures taken to aid in this process are as follows:
- Botulinum toxin
- Ablative pallidotomy or thalamotomy which are both surgical procedures to help suppress symptoms
- Deep brain stimulation
- Oral doses of drugs that help to relax muscles such as baclofen and trihexyphenidyl[13]
Epidemiology
Pantothenate kinase-associated neurodegeneration usually appears before the age of ten but about 25% of affected individuals have an onset that is atypical (after the age of ten) with a more gradual progression of the disease.[13] PKAN is very rare and is thought to be present in one to three people per one million worldwide and is not specific to any particular ethnic group.[12]
McLeod syndrome
McLeod syndrome is a neurodegenerative disease characterized by movement disorder, cognitive impairment and psychiatric symptoms. Movement in many parts of the body is impaired by this syndrome. The symptoms are mostly consistent with the general symptoms for neuroacanthocytosis disorders. Heart problems such as arrhythmia and dilated cardiomyopathy (enlarged heart) are also commonly seen in individuals with this disease. Behaviorial changes are also an early indicator of McLeod syndrome.[14]
Characteristics
McLeod syndrome is diagnosed in individuals with the following manifestations:
- McLeod blood group phenotype
- Family history of McLeod syndrome
- Central nervous system manifestations such as seizures
- Neuromuscular manifestations such as myopathy
- Dilated cardiomyopathy and arrhythmias
- Acanthocytosis [15]
Genetics
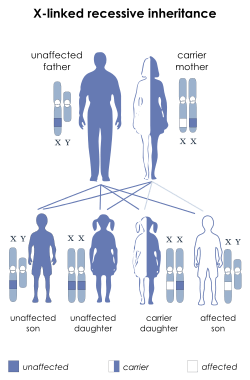
McLeod syndrome is unique in that it is inherited in an X-linked recessive manner. The mutated gene that causes McLeod syndrome is located on the X chromosome. Males (only have one X-chromosome) who have the mutation will pass their affected X chromosome to all of their daughters and to none of their sons. Males only need one mutated copy of the gene to acquire McLeod syndrome. Women (have two X-chromosomes) on the other hand, need both copies of their X-chromosomes to contain the mutated gene to be affected by the disease. Therefore, women who are carriers (heterozygous) pass their mutated X-chromosome to their offspring 50% of the time. When their sons receive the mutated chromosome, they will be affected by McLeod syndrome because they only have one X chromosome. For the daughters to become affected, they must receive a mutated chromosome from both parents. Because of this, McLeod syndrome is much more prevalent in males than it is in females.[14]
The mutated gene that causes McLeod syndrome is the XK gene located on the X-chromosome. This gene is responsible for producing the XK protein which carries the blood antigen Kx (Kell antigen). Mutations of this gene result in an abnormally short, nonfunctional form of the protein XK which leads to an absence of Kx antigens on red blood cells. This absence of the Kx antigen leads to the blood phenotype known as the "McLeod phenotype". While the absence of the Kx antigen potentially causes many blood problems for the affected individual, such as potential blood transfusions, it remains unknown how the mutation of the XK gene leads to the other problems present in individuals with McLeod syndrome.[14]
Management
Various medicines are prescribed to patients such as;
- Dopamine antagonists are prescribed to patients to suppress involuntary movement
- Antipsychotics are prescribed for psychiatric problems
- Anti-epileptic drugs for treating seizures[15]
Epidemiology
The appearance of symptoms in McLeod syndrome usually appear in early to mid-adulthood beginning with behavioral changes and movement difficulties that worsen with age.[14] McLeod syndrome is very rare. There are approximately 150 cases of McLeod syndrome worldwide and it is much more prevalent in males due to its mode of inheritance.[14]
Huntington's disease-like 2
Huntington’s disease-like 2 syndrome is a disease that resembles Huntington's disease and that occurs in people with characteristic features of Huntington's disease without the mutation in the gene associated with the disorder, HD. It belongs to a family of four Huntington’s disease-like syndromes. Huntington’s disease-like 2 is a neurodegenerative disease that affects movement, cognitive and emotional impairment. It is consistent with many of the general symptoms of neuroacanthocytosis.[16]
Characteristics
Molecular genetic testing (DNA tests) must be done to diagnose a person with Huntington’s disease-like 2 because clinical diagnosis is not enough. Individuals that are to be tested usually have symptoms that are similar to Huntington’s disease or who have a family history of Huntington’s disease-like syndromes.[17]
Genetics

Huntington’s disease-like 2 is inherited in an autosomal dominant manner. Because it is autosomal dominant, only one copy of the gene needs to be mutated for the individual to be affected by the disease. At least one parent must be affected to have an affected child.If both parents are heterozygous (each have one affected copy of the gene and one normal copy) then there is a 75% chance that their children will acquire the disease.[17]
The gene responsible for this disease is JPH3 which is located on chromosome 16q24.3. JPH3 is responsible for making the protein junctophilin-3. This protein is found in the brain and is thought to help form the junctional membrane complex. This complex is thought to be involved in the release of charged calcium ions, which is crucial in cell-cell signaling, in this case specifically, neuron signaling within the brain. The mutation of this gene is a series of CTG/CAG trinucleotide repeats causing a malfunctioning gene. A normal individual usually has between 6 and 27 of these repeats in the JPH3 gene. However, an individual affected with Huntington's disease-like 2 has between 41 and 58 trinucleotide repeats.[18]
Prevention
There are no known prevention methods to slow the progression of HDL2. Certain measures should be taken to prevent falls and other injuries that may be common due to the movement difficulties for the affected individual. Examples of this are removing items such as loose rugs and clutter from the individual's space and monitoring nutrition and swallowing to prevent aspiration.[17]
Management
Because Huntington’s disease-like 2 is very rare, treatment is solely based and built upon the treatments for Huntington’s disease and other neurodegenerative disorders. The main forms of treatment are;
- Medications to suppress abnormal movements
- Antidepressants, antipsychotics, and mood stimulants are used to treat the psychiatric manifestations
- Education about the disease can reduce stress and guilt in the patient and family[17]
Epidemiology
Huntington’s disease-like 2 usually appears around midlife but anticipation (becoming progressively worse as it is passed from generation to generation) is expected. An affected individual may live 10–20 years after onset of symptoms.[17] Huntington’s disease-like 2 is very rare, fewer than 25 pedigrees and 40 affected individuals have been discovered. The syndrome is most prevalent in South Africans for unknown reasons.[17]
History
Neuroacanthocytosis was first identified in 1950 as Bassen-Kornzweig disease, or Bassen-Kornzweig Syndrome, a rare congenital disorder in which the body failed to produce chylomicrons, a low density lipoprotein (LDL) and very low density lipoprotein (VLDL). Individuals with this condition are unable to properly digest fats. Symptoms include ataxia, peripheral neuropathy and other forms of nerve dysfunction. It was first noted by the North American physician Frank Bassen, who later partnered with the ophthalmologist Abraham Kornzweig to identify and describe causes and symptoms of the disease.[19]
Characteristics of the syndrome includes the presence of acanthocytes (burr-cell malformation of the erythrocytes), and the reduction or even absence of B-lipoproteins. Complications include retinitis pigmentosa,[20] degenerative changes in the central nervous system involving the cerebellum and long tracts, fatty diarrhea, ataxia, areflexia, demyelination, defective intestinal lipid absorption with low serum cholesterol level, intestinal malabsorption, amaurosis, retarded growth, and steatorrhea. Intellectual development may also be slightly retarded.[21] Many afflicted with the syndrome are unable to walk.[22] The syndrome appears in infancy. Affected children appear normal at birth but usually fail to thrive during their first year. The syndrome predominates in males (71%). Most cases occur in children of Jewish descent, especially among Ashkenazi Jews. The disease is transmitted in an autosomal recessive manner.[21] It is also commonly recognized as abetalipoprotein deficiency[23] or abetalipoproteinemia.[22]
A second form of neuroacanthocytosis, Levine-Critchley syndrome, was discovered by the American internist Irvine M. Levine in 1960. He described in Neurology in 1964, and again in 1968.[24] Subsequently, similar symptoms were identified and described by the British neurologist MacDonald Critchley in 1968.[25] In both cases, the physicians described a hereditary syndrome that combined acanthocytosis with neurological peculiarities but normal serum lipoprotein. Various patterns of neurologic conditions were similar to Gille de la Tourette’s syndrome, Huntington’s and chorea syndromes, Friedreich’s syndrome, and parkinsonism. Specific symptoms included tics, grimacing, movement disorders, difficulty swallowing, poor coordination, hyporeflexia, chorea, and seizures. Patients often mutilated their tongues, lips, and cheeks. The diseases appeared in both sexes, and were usually diagnosed in infancy.[26]
Research
There is research being done by the NINDS to increase scientific understanding of these disorders as well to identify prevention and treatment methods. The genetic mutations of some of the diseases have been identified and are being studied as well. Researchers suspect that the basal ganglia may play a role in the symptoms of chorea.[1] The Advocacy for neuroacanthocytosis patients and leading medical schools co-sponsor international symposia annually to also aid in furthering the research that is being done. These symposiums are all endorsed by the Movement Disorder Society and various other sponsors. The first symposium was held in 2002 and organized by a specialist in neuroacanthocytosis, Professor Adrian Danek in Seeon, Bavaria.[27]
References
Citations and notes
- ↑ 1.0 1.1 1.2 1.3 1.4 "Neuroacanthocytosis Information Page." National Institute of Neurological Disorders and Stroke (NINDS). 16 March 2009. 7 February 2010.
- ↑ Reiss, Ulrike M., Pedro A. De Alacron, and Frank E. Shafer. "Acanthocytosis: eMedicine Pediatrics: General Medicine." EMedicine - Medical Reference (2010). 7 August 2008. 8 February 2010.
- ↑ 3.0 3.1 Robertson Jr., William C., Ismeal Mohamed, and Bhagwan Moorjani. "Chorea In Children." EMedicine - Medical Reference (2008). 23 September 2008. 8 February 2010.
- ↑ Rampoldi L, Danek A, Monaco AP (2002). "Clinical features and molecular bases of neuroacanthocytosis". Journal of Molecular Medicine 80 (8): 475–91. doi:10.1007/s00109-002-0349-z. PMID 12185448.
- ↑ 5.0 5.1 Rauschkolb, Paula K., and Stephen A. Berman. "Neuroacanthocytosis." EMedicine - Medical Reference (2010). 20 January 2010. 8 February 2010.
- ↑ 6.0 6.1 6.2 "Neuroacanthocytosis." Medpedia. 3 March 2010.
- ↑ "Hirnschrittmachertherapie bei Neuroakanthozytose-erster Patient erfolgreich in Deutschland behandelt" Informationsdienst Wissenschaft (German website, "science information services"), Informationsdienst Wissenschaft. February, 2013.
- ↑ Pepper J, Zrinzo L, Mirza B, Foltynie T, Limousin P, Hariz M (2013). "The Risk of Hardware Infection in Deep Brain Stimulation Surgery Is Greater at Impulse Generator Replacement than at the Primary Procedure". Stereotactic and Functional Neurosurgery 91 (1): 56–65. doi:10.1159/000343202.
- ↑ 9.0 9.1 9.2 9.3 9.4 "Chorea Acanthocytosis." Genetics Home Reference. May 2008. 7 February 2010.
- ↑ 10.0 10.1 "Choreoacanthocytosis -- GeneReviews -- NCBI Bookshelf." National Center for Biotechnology Information. Gene Reviews. 13 October 2006. 2 March 2010.
- ↑ Dobson-Stone C, Danek A, Rampoldi L et al. (November 2002). "Mutational spectrum of the CHAC gene in patients with chorea-acanthocytosis". Eur. J. Hum. Genet. 10 (11): 773–81. doi:10.1038/sj.ejhg.5200866. PMID 12404112.
- ↑ 12.0 12.1 12.2 12.3 "Pantothenate Kinase-associated Neurodegeneration." Genetics Home Reference. October 2006. 3 March 2010.
- ↑ 13.0 13.1 13.2 13.3 "Pantothenate Kinase-Associated Neurodegeneration." National Center for Biotechnology Information. Gene Reviews. 9 January 2008. 2 March 2010.
- ↑ 14.0 14.1 14.2 14.3 14.4 "McLeod Neuroacanthocytosis Syndrome." Genetics Home Reference. May 2008. 2 March 2010.
- ↑ 15.0 15.1 "McLeod Neuroacanthocytosis Syndrome -- GeneReviews -- NCBI Bookshelf." National Center for Biotechnology Information. Gene Reviews. 26 March 2007. 7 February 2010.
- ↑ "Huntington Disease-like Syndrome." Genetics Home Reference. August 2008. 2 March 2010.
- ↑ 17.0 17.1 17.2 17.3 17.4 17.5 "Huntington Disease-Like 2." National Center for Biotechnology Information. Gene Reviews. 13 August 2009. 2 March 2010.
- ↑ Danek, Adrian, Hans H. Jung, Mariarosa A.B. Melone, Luca Rampoldi, Vania Broccoli, and Ruth H. Walker. "Neuroacanthocytosis: New Developments in a Neglected Group of Dementing Disorders." Journal of the Neurological Sciences, 229-230 (2005): 171–86. Google Scholar. 14 April 2010
- ↑ "Bassen-Kornzweig disease". Mondofacto Medical dictionary. 27 September 1977.
- ↑ Bassen, F.A.; Kornzweig, A.L. (1950). "Malformation of the erythrocytes in a case of atypical retinitis pigmentosa". Blood 5 (4): 381–7. PMID 15411425.
- ↑ 21.0 21.1 Firkin, Barry G.; Judith A. Whitworth (1989). "Malformation of the erythrocytes in a case of atypical retinitis pigmentosa". Dictionary of Medical Eponyms (Parthenon Publishing Group).
- ↑ 22.0 22.1 "Bassen Kornzweig's Disease." Twenty-first Taber's Cyclopedic Medical Dictionary, Unbound Medicine.com. 2009.
- ↑ Frank A. Bassen, M.D. (Paid Obituary). New York Times. 23 February 2003.
- ↑ Levine, I.M (1989). "A Hereditary Neurological Disease with Acanthocytosis". Neurology (Cleveland Ohio) 16: 272–. Levine, I.M; J. W. Estes; J. M. Looney (1989). "Hereditary Neurological Disease with Acanthocytosis. A new Syndrome". Archives of Neurology (Chicago) 19 (4): 403–409. doi:10.1001/archneur.1968.00480040069007. PMID 5677189.
- ↑ E. M. R. Critchley, et al. "Acanthocytosis, normolipoproteinemia and multiple tics" Postgraduate Medical Journal, Leicester, 1970, 46: 698-701.
- ↑ Ole Daniel Enersen. "Levine Critchley syndrome." Whonamedit. 2008. Accessed 26 April 2010.
- ↑ Notification of Participants. Institute for Neuroacanthocytosis. Advocacy for Neuroacanthocytosis Patients. 27 April 2010.
Bibliography
- CFP. Advocacy. Institute for Neuroacanthocytosis. Advocacy for Neuroacanthocytosis Patients. 27 April 2010.
- Bassen F A, Kornzweig A L. "Malformation of the erythrocytes in a case of atypical retinitis pigmentosa". Blood 5 (381-7): 1950.
- "Chorea Acanthocytosis." Genetics Home Reference. May 2008. 7 February 2010.
- "Choreoacanthocytosis NCBI Bookshelf." National Center for Biotechnology Information. Gene Reviews, 13 October 2006. 2 March 2010.
- Critchley E.M.R. et al. (1970). ": "Acanthocytosis, normolipoproteinemia and multiple tics". Postgraduate Medical Journal 46: 698–701. doi:10.1136/pgmj.46.542.698.
- Danek Adrian, Jung Hans H., Melone Mariarosa A.B., Rampoldi Luca, Broccoli Vania, Walker Ruth H. (2005). "Neuroacanthocytosis: New Developments in a Neglected Group of Dementing Disorders". Journal of the Neurological Sciences. 229–230: 171–86.
- "Chorea Acanthocytosis." Genetics Home Reference. May 2008. 7 February 2010.
- Dobsen-Stone C (November 2002). "Mutational spectrum of the CHAC gene in patients with chorea-acanthocytosis". European Journal of Human Genetics 10 (11): 773–81. doi:10.1038/sj.ejhg.5200866. PMID 12404112.
- Firkin, Barry G. and Judith A. Whitworth. "Bassen-Kornzweig Syndrome." "Abraham Kornzweig." "Frank Bassen." Dictionary of Medical Eponyms. The Parthenon Publishing Group. 1989. New edition in 2002.
- "Huntington Disease-Like 2." National Center for Biotechnology Information. Gene Reviews, 13 August 2009. 2 March 2010.
- "Huntington Disease-like Syndrome." Genetics Home Reference, August 2008. 2 March 2010.
- "JPH3 - Junctophilin 3." Genetics Home Reference. August 2008. 2 March 2010.
- Levine, I.M. "A Hereditary Neurological Disease with Acanthocytosis." Neurology. Cleveland, Ohio, 1964, 16: 272.
- Levine,I.M. J. W. Estes, J. M. Looney: "Hereditary Neurological Disease with Acanthocytosis. A new Syndrome." Archives of Neurology. Chicago, 1968, 19: 403–409.
- "McLeod Neuroacanthocytosis Syndrome, NCBI Bookshelf." National Center for Biotechnology Information. Gene Reviews, 26 March 2007. 7 February. 2010.
- "McLeod Neuroacanthocytosis Syndrome." Genetics Home Reference. May 2008. 2 March 2010.
- Mondofacto Medical dictionary. "Bassen-Kornzweig disease." 27 September 1997.
- "Neuroacanthocytosis." Medpedia. 3 March 2010.
- "Neuroacanthocytosis Information Page." National Institute of Neurological Disorders and Stroke. 16 March 2009. 7 February 2010.
- "Pantothenate Kinase-associated Neurodegeneration." Genetics Home Reference. October 2006. 3 March 2010.
- "Pantothenate Kinase-Associated Neurodegeneration." National Center for Biotechnology Information. Gene Reviews. 9 January 2008. 2 March 2010.
- Rampoldi L, Danek A, Monaco AP (2002). "Clinical features and molecular bases of neuroacanthocytosis". Journal of Molecular Medicine 80 (8): 475–91. doi:10.1007/s00109-002-0349-z. PMID 12185448.
- Rauschkolb, Paula K., and Stephen A. Berman. "Neuroacanthocytosis." EMedicine - Medical Reference (2010). 20 January 2010. 8 February 2010.
- Reiss, Ulrike M., Pedro A. De Alacron, and Frank E. Shafer. "Acanthocytosis: eMedicine Pediatrics: General Medicine." EMedicine - Medical Reference. 7 August 2008. 8 February 2010.
- Robertson Jr., William C., Ismeal Mohamed, and Bhagwan Moorjani. "Chorea In Children." EMedicine - Medical Reference (2008). 23 September 2008. 8 February 2010.
- "" Informationsdienst Wissenschaft (German website, "science information services"), IDW Reference. February, 2013.
- Pepper J, Zrinzo L, Mirza B, Foltynie T, Limousin P, Hariz M (2013). "The Risk of Hardware Infection in Deep Brain Stimulation Surgery Is Greater at Impulse Generator Replacement than at the Primary Procedure". Stereotactic and Functional Neurosurgery 91 (1): 56–65. doi:10.1159/000343202.
External links
- Neuroacanthocytosis information and research. Online Newsletter.
- Neurologic Disorders Index, National Institutes of Health.
- Healthwise. CIGNA.
Chromosome disorders at the US National Library of Medicine Medical Subject Headings (MeSH)