Modern valence bond theory
Modern valence bond theory is the application of valence bond theory, with computer programs that are competitive in accuracy and economy with programs for the Hartree–Fock method and other molecular orbital based methods. The latter methods dominated quantum chemistry from the advent of digital computers because they were easier to program. The early popularity of valence bond methods thus declined. It is only recently that the programming of valence bond methods has improved. These developments are due to and described by Gerratt, Cooper, Karadakov and Raimondi (1997); Li and McWeeny (2002); Joop H. van Lenthe and co-workers (2002);[1] Song, Mo, Zhang and Wu (2005); and Shaik and Hiberty (2004).[2]
In its simplest form the overlapping atomic orbitals are replaced by orbitals which are expanded as linear combinations of the atom-based basis functions, forming linear combinations of atomic orbitals (LCAO). This expansion is optimized to give the lowest energy. This procedure gives good energies without including ionic structures.
For example, in the hydrogen molecule, classic valence bond theory uses two 1s atomic orbitals (a and b) on the two hydrogen atoms respectively and then constructs a covalent structure:-
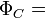 (a(1)b(2) + b(1)a(2)) (α(1)β(2) - β(1)α(2))
(a(1)b(2) + b(1)a(2)) (α(1)β(2) - β(1)α(2))
and then an ionic structure:-
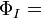 (a(1)a(2) + b(1)b(2)) (α(1)β(2) - β(1)α(2))
(a(1)a(2) + b(1)b(2)) (α(1)β(2) - β(1)α(2))
The final wave function is a linear combination of these two functions. Coulson and Fischer[3] pointed out that a completely equivalent function is:-
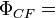 ((a+kb)(1)(b+ka)(2) + (b+ka)(1)(a+kb)(2)) (α(1)β(2) - β(1)α(2))
((a+kb)(1)(b+ka)(2) + (b+ka)(1)(a+kb)(2)) (α(1)β(2) - β(1)α(2))
as expanding this out gives a linear combination of the covalent and ionic structures. Modern valence bond theory replaces the simple linear combination of the two atomic orbitals with a linear combination of all orbitals in a larger basis set. The two resulting valence bond orbitals look like an atomic orbital on one hydrogen atom slightly distorted towards the other hydrogen atom. Modern valence bond theory is thus an extension of this Coulson–Fischer method.
Spin-coupled theory
There are a large number of different valence bond methods. Most use n valence bond orbitals for n electrons. If a single set of these orbitals is combined with all linear independent combinations of the spin functions, we have spin-coupled valence bond theory. The total wave function is optimized using the variational method by varying the coefficients of the basis functions in the valence bond orbitals and the coefficients of the different spin functions. In other cases only a sub-set of all possible spin functions is used. Many valence bond methods use several sets of the valence bond orbitals. Be warned that different authors use different names for these different valence bond methods.
Valence bond programs
Several groups have produced computer programs for modern valence bond calculations that are freely available.
References
- ↑ van Lenthe, J. H.; Dijkstra, F.; Havenith, R. W. A. TURTLE - A gradient VBSCF Program Theory and Studies of Aromaticity. In Theoretical and Computational Chemistry: Valence Bond Theory; Cooper, D. L., Ed.; Elsevier: Amsterdam, 2002; Vol. 10; pp 79--116.
- ↑ See further reading section.
- ↑ C. A. Coulson and I. Fischer, Phil. Mag. vol 40, p. 386 (1949)
Further reading
- J. Gerratt, D. L. Cooper, P. B. Karadakov and M. Raimondi, "Modern Valence Bond Theory", Chemical Society Reviews, 26, 87, 1997, and several others by the same authors.
- J. H. van Lenthe, G. G. Balint-Kurti, "The Valence Bond Self-Consistent Field (VBSCF) method", Chemical Physics Letters 76, 138–142, 1980.
- J. H. van Lenthe, G. G. Balint-Kurti, "The Valence Bond Self-Consistent Field (VBSCF) method", The Journal of Chemical Physics 78, 5699–5713, 1983.
- J. Li and R. McWeeny, "VB2000: Pushing Valence Bond Theory to new limits", International Journal of Quantum Chemistry, 89, 208, 2002.
- L. Song, Y. Mo, Q. Zhang and W. Wu, "XMVB: A program for ab initio nonorthogonal valence bond computations", Journal of Computational Chemistry, 26, 514, 2005.
- S. Shaik and P. C. Hiberty, "Valence Bond theory, its History, Fundamentals and Applications. A Primer", Reviews of Computational Chemistry, 20, 1 2004. A recent review that covers, not only their own contributions, but the whole of modern valence bond theory.