IgA nephropathy
| IgA nephropathy | |
|---|---|
 | |
| Classification and external resources | |
| ICD-10 | N02.8 |
| ICD-9 | 583.9 |
| OMIM | 161950 |
| DiseasesDB | 1353 |
| MedlinePlus | 000466 |
| eMedicine | med/886 |
| MeSH | D005922 |
IgA nephropathy (also known as IgA nephritis, IgAN, Berger disease (/bɛərˈʒeɪ/), Berger's disease, Berger's syndrome, Berger syndrome, or synpharyngitic glomerulonephritis) is a form of glomerulonephritis (inflammation of the glomeruli of the kidney).
IgA nephropathy is the most common glomerulonephritis throughout the world.[1] Primary IgA nephropathy is characterized by deposition of the IgA antibody in the glomerulus. There are other diseases associated with glomerular IgA deposits, the most common being Henoch-Schönlein purpura (HSP), which is considered by many to be a systemic form of IgA nephropathy. HSP presents with a characteristic purpuric skin rash, arthritis, and abdominal pain and occurs more commonly in young adults (16-35 yrs old). HSP is associated with a more benign prognosis than IgA nephropathy. In IgA nephropathy there is a slow progression to chronic kidney failure in 25-30% of cases during a period of 20 years.
Signs and symptoms
The classic presentation (in 40-50% of the cases) is episodic hematuria which usually starts within a day or two of a non-specific upper respiratory tract infection (hence synpharyngitic) as opposed to post-streptococcal glomerulonephritis which occurs some time (weeks) after initial infection. Less commonly gastrointestinal or urinary infection can be the inciting agent. All of these infections have in common the activation of mucosal defenses and hence IgA antibody production. Groin pain can also occur. The gross hematuria resolves after a few days, though microscopic hematuria may persist. These episodes occur on an irregular basis every few months and in most patients eventually subsides (although it can take many years). Renal function usually remains normal, though rarely, acute kidney failure may occur (see below). This presentation is more common in younger adults.
A smaller proportion (20-30%), usually the older population, have microscopic hematuria and proteinuria (less than 2 gram/day). These patients may not have any symptoms and are only clinically found if a doctor decides to take a urine sample. Hence, the disease is more commonly diagnosed in situations where screening of urine is compulsory, e.g. schoolchildren in Japan.
Very rarely (5% each), the presenting history is:
- Nephrotic syndrome (3-3.5 grams of protein loss in the urine, associated with a poorer prognosis)
- Acute kidney failure (either as a complication of the frank hematuria, when it usually recovers, or due to rapidly progressive glomerulonephritis which often leads to chronic kidney failure)
- Chronic kidney failure (no previous symptoms, presents with anemia, hypertension and other symptoms of kidney failure, in people who probably had longstanding undetected microscopic hematuria and/or proteinuria)
A variety of systemic diseases are associated with IgA nephropathy such as liver failure, celiac disease, rheumatoid arthritis, reactive arthritis, ankylosing spondylitis and HIV. Diagnosis of IgA nephropathy and a search for any associated disease occasionally reveals such an underlying serious systemic disease. Occasionally, there are simultaneous symptoms of Henoch-Schönlein purpura; see below for more details on the association. Some HLA alleles have been suspected along with complement phenotypes as being genetic factors.
Diagnosis
For an adult patient with isolated hematuria, tests such as ultrasound of the kidney and cystoscopy are usually done first to pinpoint the source of the bleeding. These tests would rule out kidney stones and bladder cancer, two other common urological causes of hematuria. In children and younger adults, the history and association with respiratory infection can raise the suspicion of IgA nephropathy. A kidney biopsy is necessary to confirm the diagnosis. The biopsy specimen shows proliferation of the mesangium, with IgA deposits on immunofluorescence and electron microscopy. However, patients with isolated microscopic hematuria (i.e. without associated proteinuria and with normal kidney function) are not usually biopsied since this is associated with an excellent prognosis. A urinalysis will show red blood cells, usually as red cell urinary casts. Proteinuria, usually less than 2 grams per day, also may be present. Other renal causes of isolated hematuria include thin basement membrane disease and Alport syndrome, the latter being a hereditary disease associated with hearing impairment and eye problems.
Other blood tests done to aid in the diagnosis include CRP or ESR, complement levels, ANA, and LDH. Protein electrophoresis and immunoglobulin levels can show increased IgA in 50% of all patients.
Morphology
Histologically, IgA nephropathy may show mesangial widening and focal and segmental inflammation. Diffuse mesangial proliferation or crescentic glomerulonephritis may also be present. Immunoflourescence shows mesangial deposition of IgA often with C3 and properdin and smaller amounts of other immunoglobulins (IgG or IgM). Early components of the classical complement pathway (C1q or C4) are usually not seen. Electron microscopy confirms electron-dense deposits in the mesangium that may extend to the subendothelial area of adjacent capillary walls in a small subset of cases, usually those with focal proliferation.
Pathophysiology
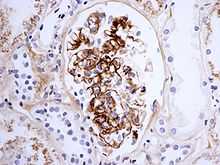
The disease derives its name from deposits of Immunoglobulin A (IgA) in a granular pattern in the mesangium (by immunofluorescence), a region of the renal glomerulus. The mesangium by light microscopy may be hypercellular and show increased deposition of extracellular matrix proteins.
There is no clear known explanation for the accumulation of the IgA. Exogenous antigens for IgA have not been identified in the kidney, but it is possible that this antigen has been cleared before the disease manifests itself. It has also been proposed that IgA itself may be the antigen.
A recently advanced theory focuses on abnormalities of the IgA1 molecule. IgA1 is one of the two immunoglobulin subclasses (the other is IgD) that is O-glycosylated on a number of serine and threonine residues in a special proline-rich hinge region. Deficiency of these sugars appears to lead to polymerisation of the IgA molecule in tissues, especially the glomerular mesangium. A similar mechanism has been claimed to underlie Henoch-Schönlein purpura (HSP), a vasculitis that mainly affects children and can feature renal involvement that is almost indistinguishable from IgA nephritis. However, human studies have found that degalactosylation of IgA1 occurs in patients with IgA nephropathy in response only to gut antigen exposures (not systemic), and occurs in healthy people to a lesser extent.[2] This strongly suggests degalactosylation of IgA1 is a result of an underlying phenomenon (abnormal mucosal antigen handling) and not the ultimate cause of IgA nephropathy. Prevailing evidence suggests that both galactose-deficient o-glycans in the hinge region of IgA1 and synthesis and binding of antibodies against IgA1 are required for immunoglobulin complexes to form and accumulate in glomeruli.[3]
From the fact that IgAN can recur after renal transplant it can be postulated that the disease is caused by a problem in the immune system rather than the kidney itself. Remarkably, the IgA1 that accumulates in the kidney does not appear to originate from the mucosa-associated lymphoid tissue (MALT), which is the site of most upper respiratory tract infections, but from the bone marrow. This, too, suggests an immune pathology rather than direct interference by outside agents.
Natural history
Since IgA nephropathy commonly presents without symptoms through abnormal findings on urinalysis, there is considerable possibility for variation in any population studied depending upon the screening policy. Similarly, the local policy for performing kidney biopsy assumes a critical role; if it is a policy to simply observe patients with isolated bloody urine, a group with a generally favourable prognosis will be excluded. If, in contrast, all such patients are biopsied, then the group with isolated microscopic hematuria and isolated mesangial IgA will be included and ‘improve’ the prognosis of that particular series.
Nevertheless, IgA nephropathy, which was initially thought to be a benign disease, has been shown to have not-so-benign long term outcomes. Though most reports describe IgA nephropathy as having an indolent evolution towards either healing or renal damage, a more aggressive course is occasionally seen associated with extensive crescents, and presenting as acute kidney failure. In general, the entry into chronic kidney failure is slow as compared to most other glomerulonephritides – occurring over a time scale of 30 years or more (in contrast to the 5 to 15 years in other glomerulonephritides). This may reflect the earlier diagnosis made due to frank hematuria.
Complete remission, i.e. a normal urinalysis, occurs rarely in adults, in about 5% of cases. Thus, even in those with normal renal function after a decade or two, urinary abnormalities persist in the great majority. In contrast, 30 – 50% of children may have a normal urinalysis at the end of 10 years. However, given the very slow evolution of this disease, the longer term (20 – 30 years) outcome of such patients is not yet established.
Overall, though the renal survival is 80–90% after 10 years, at least 25% and maybe up to 45% of adult patients will eventually develop end stage renal disease.
Therapy
The ideal treatment for IgAN would remove IgA from the glomerulus and prevent further IgA deposition. This goal still remains a remote prospect. There are a few additional caveats that have to be considered while treating IgA nephropathy. IgA nephropathy has a very variable course, ranging from a benign recurrent hematuria up to a rapid progression to chronic kidney failure. Hence the decision on which patients to treat should be based on the prognostic factors and the risk of progression. Also, IgA nephropathy recurs in transplants despite the use of ciclosporin, azathioprine or mycophenolate mofetil and steroids in these patients. There are persisting uncertainties, due to the limited number of patients included in the few controlled randomized studies performed to date, which hardly produce statistically significant evidence regarding the heterogeneity of IgA nephropathy patients, the diversity of study treatment protocols, and the length of follow-up.
Patients with isolated hematuria, proteinuria < 1 g/day and normal renal function have a benign course and are generally just followed up annually. In cases where tonsillitis is the precipitating factor for episodic hematuria, tonsillectomy has been claimed to reduce the frequency of those episodes. However, it does not reduce the incidence of progressive kidney failure.[4] Also, the natural history of the disease is such that episodes of frank hematuria reduce over time, independent of any specific treatment. Similarly, prophylactic antibiotics have not been proven to be beneficial. Dietary gluten restriction, used to reduce mucosal antigen challenge, also has not been shown to preserve kidney function. Phenytoin has also been tried without any benefit.[5]
A subset of IgA nephropathy patients, who have minimal change disease on light microscopy and clinically have nephrotic syndrome, show an exquisite response to steroids, behaving more or less like minimal change disease. In other patients, the evidence for steroids is not compelling. Short courses of high dose steroids have been proven to lack benefit. However, in patients with preserved renal function and proteinuria (1-3.5 g/day), a recent prospective study has shown that 6 months regimen of steroids may lessen proteinuria and preserve renal function.[6] However, the risks of long-term steroid use have to be weighed in such cases. It should be noted that the study had 10 years of patient follow-up data, and did show a benefit for steroid therapy; there was a lower chance of reaching end-stage renal disease (renal function so poor that dialysis was required) in the steroid group. Importantly, angiotensin-converting enzyme inhibitors were used in both groups equally.
Cyclophosphamide had been used in combination with anti-platelet/anticoagulants in unselected IgA nephropathy patients with conflicting results. Also, the side effect profile of this drug, including long term risk of malignancy and sterility, made it an unfavorable choice for use in young adults. However, one recent study, in a carefully selected high risk population of patients with declining GFR, showed that a combination of steroids and cyclophosphamide for the initial 3 months followed by azathioprine for a minimum of 2 years resulted in a significant preservation of renal function.[7] Other agents such as mycophenolate mofetil, ciclosporin and mizoribine have also been tried with varying results.
A study from Mayo Clinic did show that long term treatment with omega-3 fatty acids results in reduction of progression to kidney failure, without, however, reducing proteinuria in a subset of patients with high risk of worsening kidney function.[8] However, these results have not been reproduced by other study groups and in two subsequent meta-analyses.[9][10] However, fish oil therapy does not have the drawbacks of immunosuppressive therapy. Also, apart from its unpleasant taste and abdominal discomfort, it is relatively safe to consume.
The events that tend to progressive kidney failure are not unique to IgA nephropathy and non-specific measures to reduce the same would be equally useful. These include low-protein diet and optimal control of blood pressure. The choice of the antihypertensive agent is open as long as the blood pressure is controlled to desired level. However, Angiotensin converting enzyme inhibitors and Angiotensin II receptor antagonists are favoured due to their anti-proteinuric effect.
Genetics
Though various associations have been described, no consistent pattern pointing to a single susceptible gene has been yet identified. Associations described include those with C4 null allele, factor B Bf alleles, MHC antigens and IgA isotypes. ACE gene polymorphism (D allele) is associated with progression of kidney failure, similar to its association with other causes of chronic kidney failure. However, more than 90% of cases of IgA nephropathy are sporadic, with a few large pedigrees described from Kentucky and Italy (Online 'Mendelian Inheritance in Man' (OMIM) 161950).
Prognosis
Male gender, proteinuria (especially > 2 g/day), hypertension, smoking, hyperlipidemia, older age, familial disease and elevated creatinine concentrations are markers of a poor outcome. Frank hematuria has shown discordant results with most studies showing a better prognosis, perhaps related to the early diagnosis, except for one group which reported a poorer prognosis. Proteinuria and hypertension are the most powerful prognostic factors in this group.[11]
There are certain other features on kidney biopsy such as interstitial scarring which are associated with a poor prognosis. ACE gene polymorphism has been recently shown to have an impact with the DD genotype associated more commonly with progression to kidney failure.
Epidemiology
Men are affected three times as often as women. There is also a striking geographic variation in the prevalence of IgA nephropathy throughout the world. It is the most common glomerular disease in the Far East and Southeast Asia, comprising almost half of all the patients with glomerular disease. However, it comprises only about 25% of the proportion in European and about 10% among North Americans, with African–Americans having a very low prevalence of about 2%. A confounding factor in this analysis is the existing policy of screening and use of kidney biopsy as an investigative tool. School children in Japan undergo routine urinalysis (as do Army recruits in Singapore) and any suspicious abnormality is pursued with a kidney biopsy, which might partly explain the high incidence of IgA nephropathy in those countries.
History
Heberden first described the disease in 1801 in a 5-year-old child with abdominal pain, hematuria, hematochezia, and purpura of the legs. In 1837, Johann Schönlein described a syndrome of purpura associated with joint pain and urinary precipitates in children. Eduard Henoch, a student of Schönlein's, further associated abdominal pain and renal involvement with the syndrome. Jean Berger (1930–2011), a pioneering French Nephrologist and Hinglais, in 1968, were the first to describe IgA deposition in this form of glomerulonephritis (hence, Berger’s disease).[12]
References
- ↑ D'Amico, G (1987). "The commonest glomerulonephritis in the world: IgA nephropathy.". Q J Med 64 (245): 709–727. PMID 3329736.
- ↑ Smith AC, Molyneux K, Feehally J, Barratt J (2006). "O-glycosylation of serum IgA1 antibodies against mucosal and systemic antigens in IgA nephropathy.". J Am Soc Nephro 17 (12): 3520–3528. doi:10.1681/ASN.2006060658. PMID 17093066.
- ↑ Suzuki, Hitoshi; Kiryluk, Krzysztof; Novak, Jan; Moldoveanu, Zina; Herr, Andrew; Renfrow, Matthew; Wyatt, Robert; Scolari, Francesco; Mestecky, Jiri; Gharavi, Ali; Julian, Bruce (October 1, 2011). "The Pathophysiology of IgA Nephropathy". Journal of the American Society of Nephrology 22 (10): 1795–1803. doi:10.1681/ASN.2011050464. PMID 21949093.
- ↑ Xie Y, Chen X, Nishi S, Narita I, Gejyo F (2004). "Relationship between tonsils and IgA nephropathy as well as indications of tonsillectomy". Kidney Int. 65 (4): 1135–44. doi:10.1111/j.1523-1755.2004.00486.x. PMID 15086452.
- ↑ Clarkson AR, Seymour AE, Woodroffe AJ, McKenzie PE, Chan YL, Wootton AM (1980). "Controlled trial of phenytoin therapy in IgA nephropathy". Clin. Nephrol. 13 (5): 215–8. PMID 6994960.
- ↑ Kobayashi Y, Hiki Y, Kokubo T, Horii A, Tateno S (1996). "Steroid therapy during the early stage of progressive IgA nephropathy. A 10-year follow-up study". Nephron 72 (2): 237–42. doi:10.1159/000188848. PMID 8684533.
- ↑ Ballardie FW, Roberts IS (2002). "Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy". J. Am. Soc. Nephrol. 13 (1): 142–8. PMID 11752031.
- ↑ Donadio JV, Bergstralh EJ, Offord KP, Spencer DC, Holley KE (1994). "A controlled trial of fish oil in IgA nephropathy. Mayo Nephrology Collaborative Group". N. Engl. J. Med. 331 (18): 1194–9. doi:10.1056/NEJM199411033311804. PMID 7935657.
- ↑ Strippoli GF, Manno C, Schena FP (2003). "An "evidence-based" survey of therapeutic options for IgA nephropathy: assessment and criticism". Am. J. Kidney Dis. 41 (6): 1129–39. doi:10.1016/S0272-6386(03)00344-5. PMID 12776264.
- ↑ Dillon JJ (1997). "Fish oil therapy for IgA nephropathy: efficacy and interstudy variability". J. Am. Soc. Nephrol. 8 (11): 1739–44. PMID 9355077.
- ↑ Bartosik LP, Lajoie G, Sugar L, Cattran DC (2001). "Predicting progression in IgA nephropathy". Am. J. Kidney Dis. 38 (4): 728–35. doi:10.1053/ajkd.2001.27689. PMID 11576875.
- ↑ Berger J, Hinglais N (1968). "Les depots intercapillaires d'IgA-IgG". J Urol Nephrol 74: 694–5.
External links
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||