Hydroboration
In chemistry, hydroboration refers to the addition of a hydrogen-boron bond to C-C, C-N, and C-O double bonds, as well as C-C triple bonds. This chemical reaction is useful in the organic synthesis of organic compounds. The development of this technology and the underlying concepts were recognized by the Nobel Prize in Chemistry to Herbert C. Brown.[1] He shared the Nobel prize in chemistry with Georg Wittig in 1979 for his pioneering research on organoboranes as important synthetic intermediates.
Hydroboration produces organoborane compounds that react with a variety of reagents to produce useful compounds, such as alcohols, amines, alkyl halides. The most widely known reaction of the organoboranes is oxidation to produce alcohols typically by hydrogen peroxide. This type of reaction has promoted research on hydroboration because of its mild condition and a wide scope of olefins tolerated. Another research subtheme is metal-catalysed hydroboration.
Addition of a H-B bond to C-C double bonds
Hydroboration is typically anti-Markovnikov, i.e. the hydrogen adds to the most substituted carbon of the double bond. That the regiochemistry is reverse of a typical HX addition reflects the polarity of the Bδ+-Hδ− bonds. Hydroboration proceeds via a four-membered transition state: the hydrogen and the boron atoms added on the same face of the double bond. Granted that the mechanism is concerted, the formation of the C-B bond proceeds slightly faster than the formation of the C-H bond. As a result, in the transition state, boron develops a partially negative charge while the more substituted carbon bears a partially positive charge. This partial positive charge is better supported by the more substituted carbon.
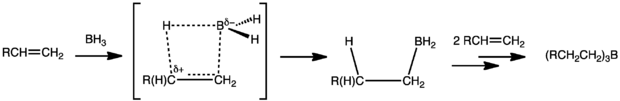
If BH3 is used as the hydroborating reagent, reactions typically proceed beyond the monoalkyl borane compounds, especially for less sterically hindered small olefins. Trisubstituted olefins can rapidly produce dialkyl boranes, but further alkylation of the organoboranes is slowed because of steric hindrance. This significant rate difference in producing di- and tri-alkyl boranes is useful in the synthesis of bulky boranes that can enhance regioselectivity (see below).
Reactions involving substituted alkenes
For trisubstituted alkenes such as 1, boron is predominantly placed on the less substituted carbon.[2]The minor product, in which the boron atom is placed on the more substituted carbon, is usually produced in less than 10%. A notable case with lower regioselectivity is styrene, and the selectivity is strongly influenced by the substituent on the para position.
.png)
Hydroboration of 1,2-disubstituted alkenes, such as a cis or trans olefin, produces generally a mixture of the two organoboranes of comparable amounts, even if the substituents are very different in terms of steric bulk. For such 1,2-disubstituted olefins, regioselectivity can be observed only when one of the two substituents is a phenyl ring. In such cases, such as trans-1-phenylpropene, the boron atom is placed on the carbon adjacent to the phenyl ring. The observations above indicate that the addition of H-B bond to olefins is under electronic control rather than steric control.
-prop-1-en-1-ylbenzene.png)
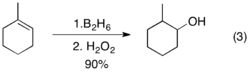
Reactions of Organoboranes
The C-B bonds generated by hydroboration are reactive with various reagents, the most common one being hydrogen peroxide. Because the addition of H-B to olefins is stereospecific, this oxidation reaction will be diastereoselective when the alkene is tri-substituted.[3] Hydroboration-oxidation is thus an excellent way of producing alcohols in a stereospecific and anti-Markovnikov fashion.

Hydroboration can also lead to amines by treating the intermediate organoboranes with chloramine or O-hydroxylaminesulfonic acid (HSA).[4]
Terminal olefins are converted to the corresponding alkyl bromides and alkyl iodides by treating the organoborane intermediates with bromine[5] or iodine.[6] Such reactions have not however proven very popular, because succinimide-based reagents such as NIS and NBS are more versatile and do not require rigorous conditions as do organoboranes.
Borane sources
Of the many hydroborating reagents available, borane (BH3) is commercially available as THF solutions wherein it exists as the adduct BH3(THF). Long term storage of BH3/THF requires stabilization by a small amount of sodium borohydride and storage at 0 °C. The concentration of BH3 usually cannot exceed 2M.[7] The related borane dimethylsulfide complex BH3S(CH3)2 (BMS) is comparatively more convenient because it is more stable[8] and it can be obtained in highly concentrated forms.[9] Less volatile sulfides have also been developed for odor control. These borane sulfide adducts are stable at room temperature and soluble in ethers and dichloromethane.
Borane adducts with phosphines and amines are also available.[10] Borane makes a strong adduct with triethylamine; using this adduct require harsher condition in hydroboration. This can be advantageous for cases such as hydroborating trienes to avoid polymerization. More sterically hindered tertiary and silyl amines can deliver borane to alkenes at room temperature. A few examples are shown below. Another advantage of these borane complexes is that it is possible to recover the amine carriers.
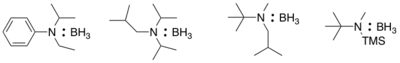
Monosubstituted boranes
Monosubstituted boranes of the form RBH2 are available for R = alkyl and halide. One important example of monoalkyl boranes is IpcBH2, monoisopinocampheylborane. It is available in both enantiomeric forms. Monobromo- and monochloro-borane can be prepared from BMS and the corresponding boron trihalides. The stable complex of monochloroborane and 1,4-dioxane is a superior for selective hydroboration of terminal alkenes.[11]
Disubstituted boranes
Hydroboration can be highly regio- and diastereoselective by using bulky dialkylborane compounds such as bis-3-methyl-2-butylborane (disiamylborane). Another dialkylborane that exhibits excellent selectivity is 9-borabycyclo[3,3,1]nonane, commonly abbreviated as 9-BBN.[12] Relative to disiamylborane, 9-BBN is more sensitive to electronic influences. Additionally, 9-BBN allows shorter reaction time and higher regioselectivity.
Disiamylborane
Among hindered dialkylboranes, disiamylborane (abbreviated Sia2BH) is well known for selective hydroboration of less hindered, usually terminal alkenes in the presence of more substituted alkenes.[13] Disiamylborane must be freshly prepared as its solutions can only be stored at 0 °C for a few hours. Dicyclohexylborane, Chx2BH, is another example that has improved thermal stability than Sia2BH.
9-BBN
The most versatile among dialkylboranes is 9-BBN. It exists predominantly as a dimer. It can be distilled without decomposition at 195 °C (12mm Hg). Such property allows 9-BBN to react at 60–80 °C, and most alkenes react within one hour in such temperature range. Even tetrasubstituted alkenes undergo hydroboration with 9-BBN at elevated temperature. As mentioned before, 9-BBN has excellent regioselectivity in hydroboration of alkenes. It is more sensitive to subtle steric differences than Sia2BH, because the rigid hetereocyclic substituents prevent internal rotation to relieve steric hindrance in the transition state. Reflecting its greater sensitivity to electronic factors, 8fu-BBN is more reactive towards alkenes than alkynes.[14]
Other secondary boranes
Simple, unhindered dialkylboranes are reactive at room temperature towards most alkenes and terminal alkynes but are difficult to prepare in high purity, since they exist in equilibrium with mono- and trialkylboranes. One common way of preparing them is the reduction of dialkylhalogenoboranes with metal hydrides.[15] An important synthetic application using such dialkylboranes, such as diethylborane, is the transmetallation of the organoboron compounds to form organozinc compounds. [16] [17] Dimesitylborane (C6H2Me3)2BH is a particularly bulky secondary borane. Because of severe steric hindrance, it does not react readily even with simple terminal alkenes. Prolonged reaction time is required at room temperature. On the other hand, alkynes undergo monohydroboration with Mes2BH easily to produce alkenylboranes.[18]
For catalytic hydroboration, Pinacol pinacolborane and catecholborane are widely used. They also demonstrate higher reactivity toward alkynes.[19]
References
- ↑ Brown, H.C. (1961). "Hydroboration-A Powerful Synthetic Tool". Tetrahedron 12: 117. doi:10.1016/0040-4020(61)80107-5.
- ↑ Brown, H. C.; Zwefei, G. (1960). "Isomerization of Organoboranes Derived Addition Mechanism of Isomerization from Branched-Chain and Ring Olefins- Further Evidence for the Elimination-Addition Mechanism of Isomerizaton". Journal of the American Chemical Society 82: 1504. doi:10.1021/ja01491a058.
- ↑ Allred, E. L.; Sonnenbcrg, J.; Winstcin S. (1960). "Preparation of Homobenzyl and Homoallyl Alcohols by the Hydroboration Method". Journal of Organic Chemistry 25: 25. doi:10.1021/jo01071a007.
- ↑ Hydroxylamine
- ↑ Brown, H. C.; Lane, C. F. (1970). "The Base-Induced Reaction of Organoboranes with Bromine. A Convenient Procedure for the Anti-Markovnikov Hydrobromination of Terminal Olefins via Hydroboration-Bromination". Journal of the American Chemical Society 92: 6660. doi:10.1021/ja00725a057.
- ↑ Brown, H. C.; Rathke, M.; Rogic, M. M. (1968). "A Fast Reaction of Organoboranes with Iodine under the Influence of Base. A Convenient Procedure for the Conversion of Terminal Olefins into Primary Iodides via Hydroboration-Iodination". Journal of the American Chemical Society 90: 5038. doi:10.1021/ja01020a056.
- ↑ See Borane-dimethylsulfide complex
- ↑ Kollonitisch, J (1961). "Reductive Ring Cleavage of Tetrahydrofurans by Diborane". J. Am. Chem. Soc. 83: 1515. doi:10.1021/ja01467a056.
- ↑ Marek Zaidlewicz, Ofir Baum, Morris Srebnik, "Borane Dimethyl Sulfide" Encyclopedia of Reagents for Organic Synthesis doi:10.1002/047084289X.rb239.pub2
- ↑ Carboni, B.; Mounier, L. (1999). "Recent developments in the chemistry of amine- and phosphine-boranes". Tetrahedron 55: 1197. doi:10.1016/S0040-4020(98)01103-X.
- ↑ Kanth, J. V. B.; Brown, H.C. (2001). "Hydroboration. 97. Synthesis of New Exceptional Chloroborane−Lewis Base Adducts for Hydroboration. Dioxane−Monochloroborane as a Superior Reagent for the Selective Hydroboration of Terminal Alkenes". Journal of Organic Chemistry 66: 5359. doi:10.1021/jo015527o.
- ↑ Dhillon, R. S. (2007). Hydroboration and organic synthesis : 9-Borabicyclo [3.3.1] Nonane (9-BBN). Springer.
- ↑ Dodd, D.S.; Ochlschlager, A. C.; (1992). "Synthesis of inhibitors of 2,3-oxidosqualene-lanosterol cyclase: conjugate addition of organocuprates to N-(carbobenzyloxy)-3-carbomethoxy-5,6-dihydro-4-pyridone". Journal of Organic Chemistry 57: 2794. doi:10.1021/jo00036a008.
- ↑ Brown, C. A.; Coleman, R. A. (1979). "Selective hydroboration of double bonds in the presence of triple bonds by 9-borabicyclo[3.3.1]nonane. A new route to acetylenic organoboranes and alcohols". Journal of Organic Chemistry 44: 2328. doi:10.1021/jo01327a079.
- ↑ Brown, H. C.; Kulkarni, S. U.; (1981). "Organoboranes: XXV. Hydridation of dialkylhaloboranes. New practical syntheses of dialkylboranes under mild conditions". Journal of Organometallic Chemistry 218: 299. doi:10.1016/S0022-328X(00)81001-3.
- ↑ Boudier, A.; Hupe, E.; Knochel, P. (2000). "Highly Diastereoselective Synthesis of Monocyclic and Bicyclic Secondary Diorganozinc Reagents with Defined Configuration". Angewandte Chemie International Edition 39: 2294. doi:10.1002/1521-3773(20000703)39:13.
- ↑ Hupe, E.; Knochel, P. (2001). "Stereoselective Synthesis of Secondary Organozinc Reagents and Their Reaction with Heteroatomic Electrophiles". Organic Letters 3: 127. doi:10.1021/ol0068400.
- ↑ Pelter, A.; Singaram, S.; Brown, H. C. (1983). "The dimesitylboron group in organic chemistry. 6 Hydroborations with dimesitylborane". Tetrahedron Letters 24: 1433. doi:10.1016/S0040-4039(00)81675-5.
- ↑ Brown, H.C.; Zaidlewicz, M. (2001). Organic Syntheses Via Boranes, Vol. 2. Milwaukee, WI: Aldrich Chemical Co.,. ISBN 978-0-9708441-0-1.