Human genetic variation


Human genetic variation is the genetic differences both within and among populations. There may be multiple variants of any given gene in the human population (genes), leading to polymorphism. Many genes are not polymorphic, meaning that only a single allele is present in the population: the gene is then said to be fixed.[1] On average, biochemically all humans are 99.5% similar to any other humans.[2]
No two humans are genetically identical. Even monozygotic twins, who develop from one zygote, have infrequent genetic differences due to mutations occurring during development and gene copy-number variation.[3] Differences between individuals, even closely related individuals, are the key to techniques such as genetic fingerprinting. Alleles occur at different frequencies in different human populations, with populations that are more geographically and ancestrally remote tending to differ more.
Causes of differences between individuals include the exchange of genes during meiosis and various mutational events. There are at least two reasons why genetic variation exists between populations. Natural selection may confer an adaptive advantage to individuals in a specific environment if an allele provides a competitive advantage. Alleles under selection are likely to occur only in those geographic regions where they confer an advantage. The second main cause of genetic variation is due to the high degree of neutrality of most mutations. Most mutations do not appear to have any selective effect one way or the other on the organism. The main cause is genetic drift, this is the effect of random changes in the gene pool. In humans, founder effect and past small population size (increasing the likelihood of genetic drift) may have had an important influence in neutral differences between populations. The theory that humans recently migrated out of Africa supports this.
The study of human genetic variation has both evolutionary significance and medical applications. It can help scientists understand ancient human population migrations as well as how different human groups are biologically related to one another. For medicine, study of human genetic variation may be important because some disease-causing alleles occur more often in people from specific geographic regions. New findings show that each human has on average 60 new mutations compared to their parents.[4][5] Apart from mutations, many genes that may have aided humans in ancient times plague humans today. For example, it is suspected that genes that allow humans to more efficiently process food are those that make people susceptible to obesity and diabetes today.[6]
Measures of variation
Genetic variation among humans occurs on many scales, from gross alterations in the human karyotype to single nucleotide changes.[7]
Nucleotide diversity is the average proportion of nucleotides that differ between two individuals. The human nucleotide diversity is estimated to be 0.1%[8] to 0.4% of base pairs.[9] A difference of 1 in 1,000 amounts to approximately 3 million nucleotide differences, because the human genome has about 3 billion nucleotides.
Single nucleotide polymorphisms
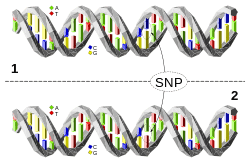
A single nucleotide polymorphism (SNP) is difference in a single nucleotide between members of one species that occurs in at least 1% of the population. It is estimated that there are 10 to 30 million SNPs in humans.
SNPs are the most common type of sequence variation, estimated to account for 90% of all sequence variation.[10] Other sequence variations are single base exchanges, deletions and insertions.[10] SNPs occur on average about every 100 to 300 bases [10] and so are the major source of heterogeneity.
A functional, or non-synonymous, SNP is one that affects some factor such as gene splicing or messenger RNA, and so causes a phenotypic difference between members of the species. About 3% to 5% of human SNPs are functional (see International HapMap Project). Neutral, or synonymous SNPs are still useful as genetic markers in genome-wide association studies, because of their sheer number and the stable inheritance over generations.[10]
A coding SNP is one that occurs inside a gene. There are 105 Human Reference SNPs that result in premature stop codons in 103 genes. This corresponds to 0.5% of coding SNPs. They occur due to segmental duplication in the genome. These SNPs result in loss of protein, yet all these SNP alleles are common and are not purified in negative selection.[11]
Structural variation
Structural variation is the variation in structure of an organism's chromosome. Structural variations, such as copy-number variation and deletions, inversions, insertions and duplications, account for much more human genetic variation than single nucleotide diversity. This was concluded in 2007 from analysis of the diploid full sequences of the genomes of two humans: Craig Venter and James D. Watson. This added to the two haploid sequences which were amalgamations of sequences from many individuals, published by the Human Genome Project and Celera Genomics respectively.[12]
Copy number variation
A copy-number variation (CNV) is a difference in the genome due to deleting or duplicating large regions of DNA on some chromosome. It is estimated that 0.4% of the genomes of unrelated humans differ with respect to copy number. When copy number variation is included, human-to-human genetic variation is estimated to be at least 0.5% (99.5% similarity).[13][14][15][16] Copy number variations are inherited but can also arise during development.[17][18][19][20]
Epigenetics
Epigenetic variation is variation in the chemical tags that attach to DNA and affect how genes get read. The tags, "called epigenetic markings, act as switches that control how genes can be read."[21] At some alleles, the epigenetic state of the DNA, and associated phenotype, can be inherited across generations of individuals.[22]
Genetic variability
Genetic variability is a measure of the tendency of individual genotypes in a population to vary (become different) from one another. Variability is different from genetic diversity, which is the amount of variation seen in a particular population. The variability of a trait is how much that trait tends to vary in response to environmental and genetic influences.
Clines
In biology, a cline is a continuum of species, populations, races, varieties, or forms of organisms that exhibit gradual phenotypic and/or genetic differences over a geographical area, typically as a result of environmental heterogeneity.[23][24][25] In the scientific study of human genetic variation, a gene cline can be rigorously defined and subjected to quantitative metrics.
Haplogroups
In the study of molecular evolution, a haplogroup is a group of similar haplotypes that share a common ancestor with a single nucleotide polymorphism (SNP) mutation. Haplogroups pertain to deep ancestral origins dating back thousands of years.[26]
The most commonly studied human haplogroups are Y-chromosome (Y-DNA) haplogroups and mitochondrial DNA (mtDNA) haplogroups, both of which can be used to define genetic populations. Y-DNA is passed solely along the patrilineal line, from father to son, while mtDNA is passed down the matrilineal line, from mother to both daughter and son. The Y-DNA and mtDNA may change by chance mutation at each generation.
Variable number tandem repeats
A variable number tandem repeat (VNTR) is the variation of length of a tandem repeat. A tandem repeat is the adjacent repetition of a short nucleotide sequence. Tandem repeats exist on many chromosomes, and their length varies between individuals. Each variant acts as an inherited allele, so they are used for personal or parental identification. Their analysis is useful in genetics and biology research, forensics, and DNA fingerprinting.
Short tandem repeats (about 5 base pairs) are called microsatellites, while longer ones are called minisatellites.
History and geographic distribution
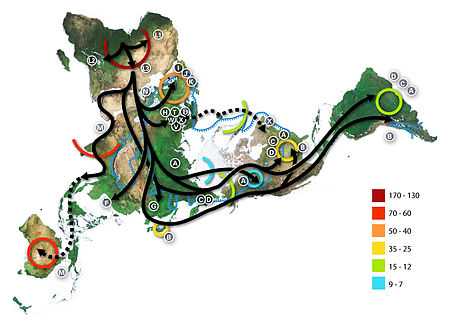
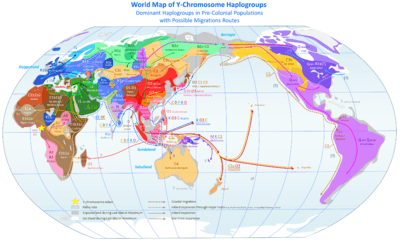
The Out of Africa theory (more precisely called "recent African origin of modern humans") is the most widely accepted explanation of the origin and early dispersal of anatomically modern humans, Homo sapiens sapiens. The theory states that archaic Homo sapiens evolved into modern humans solely in Africa, 200,000 to 100,000 years ago; around that time, one African subpopulation speciated when gene flow was restricted between African and Eurasian human populations; members of that subpopulation left Africa by 60,000 years ago and over time replaced earlier human populations such as Neanderthals and Homo erectus on Earth. Alternative theories include the multiregional origin of modern humans hypothesis.
The theory is supported by both genetic and fossil evidence. The hypothesis originated in the 19th century, with Darwin's Descent of Man, but remained speculative until the 1980s when it was supported by study of present-day mitochondrial DNA, combined with evidence from physical anthropology of archaic specimens. A large study published in 2009 found that modern humans probably originated near the border of Namibia and South Africa (reported as Namibia and Angola by BBC[27]), and left Africa through East Africa; Africa contains the most human genetic diversity anywhere on Earth, and the genetic structure of Africans traces to 14 ancestral population clusters that correlate with ethnicity and culture or language. The study lasted 10-years and analyzed variations at 1,327 DNA markers of 121 African populations, 4 African American populations, and 60 non-African populations.[28][29]
According to a 2000 study of Y-chromosome sequence variation,[10] human Y-chromosomes trace ancestry to Africa, and the descendants of the derived lineage left Africa and eventually were replaced by archaic human Y-chromosomes in Eurasia. The study also shows that a minority of contemporary East Africans and Khoisan are the descendants of the most ancestral patrilineages of anatomically modern humans that left Africa 35,000 to 89,000 years ago.[10] Other evidence supporting the theory is that variations in skull measurements decrease with distance from Africa at the same rate as the decrease in genetic diversity. Human genetic diversity decreases in native populations with migratory distance from Africa, and this is thought to be due to bottlenecks during human migration, which are events that temporarily reduce population size.[30][31]
Population genetics
In the field of population genetics, it is believed that the distribution of neutral polymorphisms among contemporary humans reflects human demographic history. It has been theorized that humans passed through a population bottleneck before a rapid expansion coinciding with migrations out of Africa leading to an African-Eurasian divergence around 100,000 years ago (ca. 5,000 generations), followed by a European-Asian divergence about 40,000 years ago (ca. 2,000 generations). Richard G. Klein, Nicholas Wade and Spencer Wells, among others, have postulated that modern humans did not leave Africa and successfully colonize the rest of the world until as recently as 60,000 - 50,000 years B.P., pushing back the dates for subsequent population splits as well.
The rapid expansion of a previously small population has two important effects on the distribution of genetic variation. First, the so-called founder effect occurs when founder populations bring only a subset of the genetic variation from their ancestral population. Second, as founders become more geographically separated, the probability that two individuals from different founder populations will mate becomes smaller. The effect of this assortative mating is to reduce gene flow between geographical groups, and to increase the genetic distance between groups. The expansion of humans from Africa affected the distribution of genetic variation in two other ways. First, smaller (founder) populations experience greater genetic drift because of increased fluctuations in neutral polymorphisms. Second, new polymorphisms that arose in one group were less likely to be transmitted to other groups as gene flow was restricted.
Our history as a species also has left genetic signals in regional populations. For example, in addition to having higher levels of genetic diversity, populations in Africa tend to have lower amounts of linkage disequilibrium than do populations outside Africa, partly because of the larger size of human populations in Africa over the course of human history and partly because the number of modern humans who left Africa to colonize the rest of the world appears to have been relatively low (Gabriel et al. 2002). In contrast, populations that have undergone dramatic size reductions or rapid expansions in the past and populations formed by the mixture of previously separate ancestral groups can have unusually high levels of linkage disequilibrium (Nordborg and Tavare 2002).
Many other geographic, climatic, and historical factors have contributed to the patterns of human genetic variation seen in the world today. For example, population processes associated with colonization, periods of geographic isolation, socially reinforced endogamy, and natural selection all have affected allele frequencies in certain populations (Jorde et al. 2000b; Bamshad and Wooding 2003). In general, however, the recency of our common ancestry and continual gene flow among human groups have limited genetic differentiation in our species.
Distribution of variation
The distribution of genetic variants within and among human populations are impossible to describe succinctly because of the difficulty of defining a "population," the clinal nature of variation, and heterogeneity across the genome (Long and Kittles 2003). In general, however, an average of 85% of genetic variation exists within local populations, ~7% is between local populations within the same continent, and ~8% of variation occurs between large groups living on different continents,. (Lewontin 1972; Jorde et al. 2000a). The recent African origin theory for humans would predict that in Africa there exists a great deal more diversity than elsewhere, and that diversity should decrease the further from Africa a population is sampled. Long and Kittles show that indeed, African populations contain about 100% of human genetic diversity, whereas in populations outside of Africa diversity is much reduced, for example in their population from New Guinea only about 70% of human variation is captured.
Phenotypic variation
Sub-Saharan Africa has the most human genetic diversity and the same has been shown to hold true for phenotypic diversity.[30] Phenotype is connected to genotype through gene expression. Genetic diversity decreases smoothly with migratory distance from that region, which many scientists believe to be the origin of modern humans, and that decrease is mirrored by a decrease in phenotypic variation. Skull measurements are an example of a physical attribute whose within-population variation decreases with distance from Africa.
The distribution of many physical traits resembles the distribution of genetic variation within and between human populations (American Association of Physical Anthropologists 1996; Keita and Kittles 1997). For example, ~90% of the variation in human head shapes occurs within continental groups, and ~10% separates groups, with a greater variability of head shape among individuals with recent African ancestors (Relethford 2002).
A prominent exception to the common distribution of physical characteristics within and among groups is skin color. Approximately 10% of the variance in skin color occurs within groups, and ~90% occurs between groups (Relethford 2002). This distribution of skin color and its geographic patterning — with people whose ancestors lived predominantly near the equator having darker skin than those with ancestors who lived predominantly in higher latitudes — indicate that this attribute has been under strong selective pressure. Darker skin appears to be strongly selected for in equatorial regions to prevent sunburn, skin cancer, the photolysis of folate, and damage to sweat glands (Sturm et al. 2001; Rees 2003).
A study published in 2007 found that 25% of genes showed different levels of gene expression between populations of European and Asian descent.[32][33][34][35][36] The primary cause of this difference in gene expression was thought to be SNPs in gene regulatory regions of DNA. Another study published in 2007 found that approximately 83% of genes were expressed at different levels among individuals and about 17% between populations of European and African descent.[37][38]
Archaic admixture
There is a hypothesis that anatomically modern humans interbred with Neanderthals during the Middle Paleolithic. In May 2010, the Neanderthal Genome Project presented genetic evidence that interbreeding did likely take place and that a small but significant portion of Neanderthal admixture is present in the DNA of modern Eurasians and Oceanians, and nearly absent in sub-Saharan African populations.
Between 4% and 6% of the genome of Melanesians (represented by the Papua New Guinean and Bougainville Islander) are thought to derive from Denisova hominins - a previously unknown species which shares a common origin with Neanderthals. It was possibly introduced during the early migration of the ancestors of Melanesians into Southeast Asia. This history of interaction suggests that Denisovans once ranged widely over eastern Asia.[39]
Thus, Melanesians emerge as the most archaic-admixed population, having Denisovan/Neanderthal-related admixture of ~8%.
In a study published in 2013, Jeffrey Wall from University of California studied whole sequence-genome data and found higher rates of introgression in Asians compared to Europeans.[40] Hammer et al. tested the hypothesis that contemporary African genomes have signatures of gene flow with archaic human ancestors and found evidence of archaic admixture in African genomes, suggesting that modest amounts of gene flow were widespread throughout time and space during the evolution of anatomically modern humans.[41]
Categorization of the world population

New data on human genetic variation has reignited the debate about a possible biological basis for categorization of humans into races. Most of the controversy surrounds the question of how to interpret the genetic data and whether conclusions based on it are sound. Some researchers argue that self-identified race can be used as an indicator of geographic ancestry for certain health risks and medications.
Although the genetic differences among human groups are relatively small, these differences in certain genes such as duffy, ABCC11, SLC24A5, called ancestry-informative markers (AIMs) nevertheless can be used to reliably situate many individuals within broad, geographically based groupings. For example, computer analyses of hundreds of polymorphic loci sampled in globally distributed populations have revealed the existence of genetic clustering that roughly is associated with groups that historically have occupied large continental and subcontinental regions (Rosenberg et al. 2002; Bamshad et al. 2003).
Some commentators have argued that these patterns of variation provide a biological justification for the use of traditional racial categories. They argue that the continental clusterings correspond roughly with the division of human beings into sub-Saharan Africans; Europeans, Western Asians, Central Asians, Southern Asians and Northern Africans; Eastern Asians, Southeast Asians, Polynesians and Native Americans; and other inhabitants of Oceania (Melanesians, Micronesians & Australian Aborigines) (Risch et al. 2002). Other observers disagree, saying that the same data undercut traditional notions of racial groups (King and Motulsky 2002; Calafell 2003; Tishkoff and Kidd 2004[9]). They point out, for example, that major populations considered races or subgroups within races do not necessarily form their own clusters.
Furthermore, because human genetic variation is clinal, many individuals affiliate with two or more continental groups. Thus, the genetically based "biogeographical ancestry" assigned to any given person generally will be broadly distributed and will be accompanied by sizable uncertainties (Pfaff et al. 2004).
In many parts of the world, groups have mixed in such a way that many individuals have relatively recent ancestors from widely separated regions. Although genetic analyses of large numbers of loci can produce estimates of the percentage of a person's ancestors coming from various continental populations (Shriver et al. 2003; Bamshad et al. 2004), these estimates may assume a false distinctiveness of the parental populations, since human groups have exchanged mates from local to continental scales throughout history (Cavalli-Sforza et al. 1994; Hoerder 2002). Even with large numbers of markers, information for estimating admixture proportions of individuals or groups is limited, and estimates typically will have wide confidence intervals (Pfaff et al. 2004).
Genetic clustering
Genetic data can be used to infer population structure and assign individuals to groups that often correspond with their self-identified geographical ancestry. Recently, Lynn Jorde and Steven Wooding argued that "Analysis of many loci now yields reasonably accurate estimates of genetic similarity among individuals, rather than populations. Clustering of individuals is correlated with geographic origin or ancestry."[8]
Forensic anthropology
Forensic anthropologists can determine aspects of geographic ancestry (i.e. Asian, African, or European) from skeletal remains with a high degree of accuracy by analyzing skeletal measurements.[42] According to some studies, individual test methods such as mid-facial measurements and femur traits can identify the geographic ancestry and by extension the racial category to which an individual would have been assigned during their lifetime, with over 80% accuracy, and in combination can be even more accurate. However, the skeletons of persons who have recent ancestry in different geographical regions, can exhibit characteristics of more than one ancestral group, and hence cannot be identified as belonging to any single ancestral group.

Gene flow and admixture
Gene flow between two populations reduces the average genetic distance between the populations, only totally isolated human populations experience no gene flow and most populations have continuous gene flow with other neighboring populations which create the clinal distribution observed for moth genetic variation. When gene flow takes place between well-differentiated genetic populations the result is referred to as "genetic admixture".
Admixture mapping is a technique used to study how genetic variants cause differences in disease rates between population.[10] Recent admixture populations that trace their ancestry to multiple continents are well suited for identifying genes for traits and diseases that differ in prevalence between parental populations. African-American populations have been the focus of numerous population genetic and admixture mapping studies, including studies of complex genetic traits such as white cell count, body-mass index, prostate cancer and renal disease.[10]
An analysis of phenotypic and genetic variation including skin color and socio-economic status was carried out in the population of Cape Verde which has a well documented history of contact between Europeans and Africans. The studies showed that pattern of admixture in this population has been sex-biased and there is a significant interactions between socio economic status and skin color independent of the skin color and ancestry.[10] Another study shows an increased risk of graft-versus-host disease complications after transplantation due to genetic variants in human leukocyte antigen (HLA) and non-HLA proteins.[10]
Health
Differences in allele frequencies contribute to group differences in the incidence of some monogenic diseases, and they may contribute to differences in the incidence of some common diseases (Risch et al. 2002; Burchard et al. 2003; Tate and Goldstein 2004). For the monogenic diseases, the frequency of causative alleles usually correlates best with ancestry, whether familial (for example, Ellis-van Creveld syndrome among the Pennsylvania Amish), ethnic (Tay-Sachs disease among Ashkenazi Jewish populations), or geographical (hemoglobinopathies among people with ancestors who lived in malarial regions). To the extent that ancestry corresponds with racial or ethnic groups or subgroups, the incidence of monogenic diseases can differ between groups categorized by race or ethnicity, and health-care professionals typically take these patterns into account in making diagnoses.[43]
Even with common diseases involving numerous genetic variants and environmental factors, investigators point to evidence suggesting the involvement of differentially distributed alleles with small to moderate effects. Frequently cited examples include hypertension (Douglas et al. 1996), diabetes (Gower et al. 2003), obesity (Fernandez et al. 2003), and prostate cancer (Platz et al. 2000). However, in none of these cases has allelic variation in a susceptibility gene been shown to account for a significant fraction of the difference in disease prevalence among groups, and the role of genetic factors in generating these differences remains uncertain (Mountain and Risch 2004).
Neil Risch of Stanford University has proposed that self-identified race/ethnic group could be a valid means of categorization in the USA for public health and policy considerations.[44][45] While a 2002 paper by Noah Rosenberg's group makes a similar claim "The structure of human populations is relevant in various epidemiological contexts. As a result of variation in frequencies of both genetic and nongenetic risk factors, rates of disease and of such phenotypes as adverse drug response vary across populations. Further, information about a patient’s population of origin might provide health care practitioners with information about risk when direct causes of disease are unknown."[46]
Genome projects
Human genome projects are scientific endeavors that determine or study the structure of the human genome. The Human Genome Project was a landmark genome project.
See also
- Race and genetics
- Archaeogenetics
- Human evolutionary genetics
- Multiregional hypothesis
- Recent single origin hypothesis
- Isolation by distance
- Genealogical DNA test
- Y-chromosome haplogroups by populations
- Human genetic clustering
Regional:
- Genetic history of Europe
- Genetic history of South Asia
- African admixture in Europe
- Genetic history of indigenous peoples of the Americas
- Genetic history of the British Isles
Projects:
References
- ↑ When all genes are fixed within a population, so every member of the population is genetically identical, the population is said to be clonal. This occurs in species that reproduce asexually.
- ↑ Dr.Craig Venter, Aaron. "In the Genome Race, the Sequel Is Personal".
- ↑ Bruder, CEG et al. (2008). "Phenotypically Concordant and Discordant Monozygotic Twins Display Different DNA Copy-Number-Variation Profiles". The American Journal of Human Genetics 82 (3): 763–771. doi:10.1016/j.ajhg.2007.12.011.
- ↑ "We are all mutants: First direct whole-genome measure of human mutation predicts 60 new mutations in each of us". Science Daily. 13 June 2011. Retrieved 2011-09-05.
- ↑ Conrad, DF et al. (2011). "Variation in genome-wide mutation rates within and between human families". Nature Genetics 43 (7): 712–4. doi:10.1038/ng.862. PMC 3322360. PMID 21666693.
- ↑ Tishkoff, S. A., & Verrelli, B. C. (2003). "PATTERNS OF HUMAN GENETIC DIVERSITY: Implications for human evolutionary history and disease". Annual Review of Genomics and Human Genetics, 4 4 (1).
- ↑ Kidd, JM et al. (2008). "Mapping and sequencing of structural variation from eight human genomes". Nature 453 (7191): 56–64. Bibcode:2008Natur.453...56K. doi:10.1038/nature06862. PMC 2424287. PMID 18451855.
- ↑ 8.0 8.1 Jorde, LB; Wooding, SP (2004). "Genetic variation, classification and 'race'". Nature Genetics 36 (11s): S28–33. doi:10.1038/ng1435. PMID 15508000.
- ↑ 9.0 9.1 Tishkoff, SA; Kidd, KK (2004). "Implications of biogeography of human populations for 'race' and medicine". Nature Genetics 36 (11s): S21–7. doi:10.1038/ng1438. PMID 15507999.
- ↑ 10.0 10.1 10.2 10.3 10.4 10.5 10.6 10.7 10.8 10.9 Collins, F. S.; Brooks, L. D.; Chakravarti, A. (1998). "A DNA polymorphism discovery resource for research on human genetic variation". Genome research 8 (12): 1229–1231. PMID 9872978.
- ↑ Ng, P. C.; Levy, S.; Huang, J.; Stockwell, T. B.; Walenz, B. P.; Li, K.; Axelrod, N.; Busam, D. A.; Strausberg, R. L.; Venter, J. C. (2008). Schork, Nicholas J, ed. "Genetic Variation in an Individual Human Exome". PLoS Genetics 4 (8): e1000160. doi:10.1371/journal.pgen.1000160. PMC 2493042. PMID 18704161.
- ↑ Gross, L (2007). "A New Human Genome Sequence Paves the Way for Individualized Genomics". PLoS Biology 5 (10): e266. doi:10.1371/journal.pbio.0050266. PMC 1964778. PMID 20076646.
- ↑ "First Individual Diploid Human Genome Published By Researchers at J. Craig Venter Institute". J. Craig Venter Institute. 3 September 2007. Retrieved 2011-09-05.
- ↑ Levy, S et al. (2007). "The Diploid Genome Sequence of an Individual Human". PLoS Biology 5 (10): e254. doi:10.1371/journal.pbio.0050254. PMC 1964779. PMID 17803354.
- ↑ "Understanding Genetics: Human Health and the Genome". The Tech Museum of Innovation. 24 January 2008. Retrieved 2011-09-05.
- ↑ "First Diploid Human Genome Sequence Shows We're Surprisingly Different". Science Daily. 4 September 2007. Retrieved 2011-09-05.
- ↑ "Copy number variation may stem from replication misstep". EurekAlert!. 27 December 2007. Retrieved 2011-09-05.
- ↑ Lee, JA; Carvalho, CMB; Lupski, JR (2007). "A DNA Replication Mechanism for Generating Nonrecurrent Rearrangements Associated with Genomic Disorders". Cell 131 (7): 1235–47. doi:10.1016/j.cell.2007.11.037. PMID 18160035.
- ↑ Redon, R et al. (2006). "Global variation in copy number in the human genome". Nature 444 (7118): 444–54. Bibcode:2006Natur.444..444R. doi:10.1038/nature05329. PMC 2669898. PMID 17122850.
- ↑ Dumas, L et al. (2007). "Gene copy number variation spanning 60 million years of human and primate evolution". Genome Research 17 (9): 1266–77. doi:10.1101/gr.6557307. PMC 1950895. PMID 17666543.
- ↑ "Human Genetic Variation Fact Sheet". National Institute of General Medical Sciences. 19 August 2011. Retrieved 2011-09-05.
- ↑ Rakyan, V; Whitelaw, E (2003). "Transgenerational epigenetic inheritance". Current Biology 13 (1): R6. doi:10.1016/S0960-9822(02)01377-5. PMID 12526754.
- ↑ "Cline". Microsoft Encarta Premium. 2009.
- ↑ King, RC; Stansfield, WD; Mulligan, PK (2006). "Cline". A dictionary of genetics (7th ed.). Oxford University Press. ISBN 978-0195307610.
- ↑ Begon, M; Townsend, CR; Harper, JL (2006). Ecology: From individuals to ecosystems (4th ed.). Wiley-Blackwell. p. 10. ISBN 978-1405111171.
- ↑ "Haplogroup". DNA-Newbie Glossary. International Society of Genetic Genealogy. Retrieved 2012-09-05.
- ↑ Gill, V (1 May 2009). "Africa's genetic secrets unlocked". BBC World News. Retrieved 2012-09-05.
- ↑ "African Genetics Study Revealing Origins, Migration And 'Startling Diversity' Of African Peoples". Science Daily. 2 May 2009. Retrieved 2011-09-05.
- ↑ Tishkoff, SA et al. (2009). "The Genetic Structure and History of Africans and African Americans". Science 324 (5930): 1035–44. Bibcode:2009Sci...324.1035T. doi:10.1126/science.1172257. PMC 2947357. PMID 19407144.
- ↑ 30.0 30.1 "New Research Proves Single Origin Of Humans In Africa". Science Daily. 19 July 2007. Retrieved 2011-09-05.
- ↑ Manica, A; Amos, W; Balloux, F; Hanihara, T (2007). "The effect of ancient population bottlenecks on human phenotypic variation". Nature 448 (7151): 346–8. Bibcode:2007Natur.448..346M. doi:10.1038/nature05951. PMC 1978547. PMID 17637668.
- ↑ Phillips, ML (9 January 2007). "Ethnicity tied to gene expression". The Scientist. Retrieved 2011-09-05.
- ↑ Spielman, RS et al. (2007). "Common genetic variants account for differences in gene expression among ethnic groups". Nature Genetics 39 (2): 226–31. doi:10.1038/ng1955. PMC 3005333. PMID 17206142.
- ↑ Swaminathan, N (9 January 2007). "Ethnic Differences Traced to Variable Gene Expression". Scientific American. Retrieved 2011-09-05.
- ↑ Check, E (2007). "Genetic expression speaks as loudly as gene type". Nature News. doi:10.1038/news070101-8.
- ↑ Bell, L (15 January 2007). "Variable gene expression seen in different ethnic groups". BioNews.org. Retrieved 2011-09-05.
- ↑ Kamrani, K (28 February 2008). "Differences of gene expression between human populations". Anthropology.net. Retrieved 2011-09-05.
- ↑ Storey, JD et al. (2007). "Gene-Expression Variation Within and Among Human Populations". The American Journal of Human Genetics 80 (3): 502–509. doi:10.1086/512017.
- ↑ Reich, D et al. (2010). "Genetic history of an archaic hominin group from Denisova Cave in Siberia". Nature 468 (7327): 1053–60. Bibcode:2010Natur.468.1053R. doi:10.1038/nature09710. PMID 21179161.
- ↑ Wall, Jeffrey D. et al. (2013). "Higher Levels of Neanderthal Ancestry in East Asians Than in Europeans". Genetics 194: 199–209. doi:10.1534/genetics.112.148213.
- ↑ Hammer, Michael F. et al. (2011). "Genetic evidence for archaic admixture in Africa". Proceedings of the National Academy of Sciences 108 (37): 15123–15128. Bibcode:2011PNAS..10815123H. doi:10.1073/pnas.1109300108. PMC 3174671. PMID 21896735.
- ↑ "Does Race Exist?". NOVA. PBS. 15 February 2000. Retrieved 2011-09-05.
- ↑ Lu, YF; Goldstein, DB; Angrist, M; Cavalleri, G (24 July 2014). "Personalized medicine and human genetic diversity.". Cold Spring Harbor perspectives in medicine 4 (9): a008581. doi:10.1101/cshperspect.a008581. PMID 25059740.
- ↑ Tang, H et al. (2005). "Genetic Structure, Self-Identified Race/Ethnicity, and Confounding in Case-Control Association Studies". The American Journal of Human Genetics 76 (2): 268–75. doi:10.1086/427888. PMC 1196372. PMID 15625622.
- ↑ Risch, N; Burchard, E; Ziv, E; Tang, H (2002). "Categorization of humans in biomedical research: genes, race and disease". Genome Biology 3 (7): 1–12. doi:10.1186/gb-2002-3-7-comment2007. PMC 139378. PMID 12184798.
- ↑ Rosenberg, NA et al. (2002). "Genetic Structure of Human Populations". Science 298 (5602): 2381–5. Bibcode:2002Sci...298.2381R. doi:10.1126/science.1078311. PMID 12493913.
- Bibliography
- Race, Ethnicity, and Genetics Working Group (2005). "The Use of Racial, Ethnic, and Ancestral Categories in Human Genetics Research". The American Journal of Human Genetics 77 (4): 519–32. doi:10.1086/491747. PMC 1275602. PMID 16175499.
- Altmüller, J; Palmer, L; Fischer, G; Scherb, H; Wjst, M (2001). "Genomewide Scans of Complex Human Diseases: True Linkage is Hard to Find". The American Journal of Human Genetics 69 (5): 936–50. doi:10.1086/324069. PMC 1274370. PMID 11565063.
- Aoki, K (2002). "Sexual selection as a cause of human skin colour variation: Darwin's hypothesis revisited". Annals of Human Biology 29 (6): 589–608. doi:10.1080/0301446021000019144. PMID 12573076.
- Bamshad, M; Wooding, S; Salisbury, BA; Stephens, JC (2004). "Deconstructing the relationship between genetics and race". Nature Reviews Genetics 5 (8): 598–609. doi:10.1038/nrg1401. PMID 15266342. reprint-zip
- Bamshad, M; Wooding, SP (2003). "Signatures of natural selection in the human genome". Nature Reviews Genetics 4 (2): 99–111. doi:10.1038/nrg999. PMID 12560807.
- Bamshad, MJ et al. (2003). "Human Population Genetic Structure and Inference of Group Membership". The American Journal of Human Genetics 72 (3): 578–89. doi:10.1086/368061. PMC 1180234. PMID 12557124.
- Cann, RL; Stoneking, M; Wilson, AC (1987). "Mitochondrial DNA and human evolution". Nature 325 (6099): 31–6. Bibcode:1987Natur.325...31C. doi:10.1038/325031a0. PMID 3025745.
- Cardon, LR; Abecasis, GR (2003). "Using haplotype blocks to map human complex trait loci". Trends in Genetics 19 (3): 135–40. doi:10.1016/S0168-9525(03)00022-2. PMID 12615007.
- Cavalli-Sforza, LL; Feldman, MW (2003). "The application of molecular genetic approaches to the study of human evolution". Nature Genetics 33 (3s): 266–75. doi:10.1038/ng1113. PMID 12610536.
- Collins, FS (2004). "What we do and don't know about 'race', 'ethnicity', genetics and health at the dawn of the genome era". Nature Genetics 36 (11s): S13–5. doi:10.1038/ng1436. PMID 15507997.
- Collins, FS; Green, ED; Guttmacher, AE; Guyer, MS (2003). "A vision for the future of genomics research". Nature 422 (6934): 835–47. Bibcode:2003Natur.422..835C. doi:10.1038/nature01626. PMID 12695777.
- Ebersberger, I; Metzler, D; Schwarz, C; Pääbo, S (2002). "Genomewide Comparison of DNA Sequences between Humans and Chimpanzees". The American Journal of Human Genetics 70 (6): 1490–7. doi:10.1086/340787. PMC 379137. PMID 11992255.
- Edwards, AWF (2003). "Human genetic diversity: Lewontin's fallacy". BioEssays 25 (8): 798–801. doi:10.1002/bies.10315. PMID 12879450.
- Foster, MW; Sharp, RR (2004). "Opinion: Beyond race: Towards a whole-genome perspective on human populations and genetic variation". Nature Reviews Genetics 5 (10): 790–6. doi:10.1038/nrg1452. PMID 15510170.
- Foster, M et al. (1999). "The Role of Community Review in Evaluating the Risks of Human Genetic Variation Research". The American Journal of Human Genetics 64 (6): 1719–27. doi:10.1086/302415. PMC 1377916. PMID 10330360.
- Gabriel, SB et al. (2002). "The Structure of Haplotype Blocks in the Human Genome". Science 296 (5576): 2225–9. Bibcode:2002Sci...296.2225G. doi:10.1126/science.1069424. PMID 12029063.
- Harding, RM et al. (2000). "Evidence for Variable Selective Pressures at MC1R". The American Journal of Human Genetics 66 (4): 1351–61. doi:10.1086/302863. PMC 1288200. PMID 10733465.
- Gyllensten, U; Ingman, M; Kaessmann, H; Pääbo, S (2000). "Mitochondrial genome variation and the origin of modern humans". Nature 408 (6813): 708–13. doi:10.1038/35047064. PMID 11130070.
- The International Hapmap Consortium (2003). "The International HapMap Project". Nature 426 (6968): 789–96. doi:10.1038/nature02168. PMID 14685227.
- The International Hapmap Consortium (2004). "Opinion: Integrating ethics and science in the International HapMap Project". Nature Reviews Genetics 5 (6): 467–75. doi:10.1038/nrg1351. PMID 15153999.
- The International Human Genome Sequencing Consortium (2001). "Initial sequencing and analysis of the human genome". Nature 409 (6822): 860–921. doi:10.1038/35057062. PMID 11237011.
- Jorde, LB; Bamshad, M; Rogers, AR (1998). "Using mitochondrial and nuclear DNA markers to reconstruct human evolution" (PDF). BioEssays 20 (2): 126–36. doi:10.1002/(SICI)1521-1878(199802)20:2<126::AID-BIES5>3.0.CO;2-R. PMID 9631658.
- Jorde, LB et al. (2000). "The Distribution of Human Genetic Diversity: A Comparison of Mitochondrial, Autosomal, and Y-Chromosome Data". The American Journal of Human Genetics 66 (3): 979–88. doi:10.1086/302825. PMC 1288178. PMID 10712212.
- Jorde, LB; Watkins, WW; Kere, J; Nyman, D; Eriksson, AW (2000). "Gene Mapping in Isolated Populations: New Roles for Old Friends?". Human Heredity 50 (1): 57–65. doi:10.1159/000022891. PMID 10545758.
- Kaessmann, H; Heißig, D; von Haeseler, A; Pääbo, S (1999). "DNA sequence variation in a non-coding region of low recombination on the human X chromosome". Nature Genetics 22 (1): 78–81. doi:10.1038/8785. PMID 10319866.
- Kaessmann, H; Wiebe, V; Weiss, G; Pääbo, S (2001). "Great ape DNA sequences reveal a reduced diversity and an expansion in humans". Nature Genetics 27 (2): 155–6. doi:10.1038/84773. PMID 11175781.
- Keita, SOY; Kittles, RA (1997). "The Persistence of Racial Thinking and the Myth of Racial Divergence". American Anthropologist 99 (3): 534–544. doi:10.1525/aa.1997.99.3.534.
- Lewontin, RC (1972). "The apportionment of human diversity". Evolutionary Biology 6: 381–398. doi:10.1007/978-1-4684-9063-3_14.
- Marks, J (1995). Human Biodiversity: Genes, Race, and History. Aldine Transaction. ISBN 978-0-202-02033-4.
- Mountain, JL; Risch, N (2004). "Assessing genetic contributions to phenotypic differences among 'racial' and 'ethnic' groups". Nature Genetics 36 (11s): S48. doi:10.1038/ng1456. PMID 15508003.
- Pääbo, S (2003). "The mosaic that is our genome". Nature 421 (6921): 409–12. Bibcode:2003Natur.421..409P. doi:10.1038/nature01400. PMID 12540910.
- Ramachandran, S et al. (2005). "Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa". Proceedings of the National Academy of Sciences 102 (44): 15942–7. Bibcode:2005PNAS..10215942R. doi:10.1073/pnas.0507611102. PMC 1276087. PMID 16243969.
- Relethford, JH (2002). "Apportionment of global human genetic diversity based on craniometrics and skin color". American Journal of Physical Anthropology 118 (4): 393–8. doi:10.1002/ajpa.10079. PMID 12124919.
- Sankar, P; Cho, MK (2002). "Toward a New Vocabulary of Human Genetic Variation". Science 298 (5597): 1337–8. doi:10.1126/science.1074447. PMC 2271140. PMID 12434037.
- Sankar, P et al. (2004). "Genetic Research and Health Disparities". JAMA: the Journal of the American Medical Association 291 (24): 2985–9. doi:10.1001/jama.291.24.2985. PMC 2271142. PMID 15213210.
- Serre, D; Pääbo, S (2004). "Evidence for Gradients of Human Genetic Diversity Within and Among Continents". Genome Research 14 (9): 1679–85. doi:10.1101/gr.2529604. PMC 515312. PMID 15342553.
- Templeton, AR (1998). "Human Races: A Genetic and Evolutionary Perspective". American Anthropologist 100 (3): 632–650. doi:10.1525/aa.1998.100.3.632.
- Weiss, KM (1998). "Coming to Terms with Human Variation". Annual Review of Anthropology 27: 273–300. doi:10.1146/annurev.anthro.27.1.273.
- Weiss, KM; Terwilliger, JD (2000). "How many diseases does it take to map a gene with SNPs?". Nature Genetics 26 (2): 151–7. doi:10.1038/79866. PMID 11017069.
- Yu, N et al. (2003). "Low nucleotide diversity in chimpanzees and bonobos". Genetics 164 (4): 1511–8. PMC 1462640. PMID 12930756.
- Ziętkiewicz, E et al. (2003). "Haplotypes in the Dystrophin DNA Segment Point to a Mosaic Origin of Modern Human Diversity". The American Journal of Human Genetics 73 (5): 994–1015. doi:10.1086/378777. PMC 1180505. PMID 14513410.
Further reading
- Pennisi, E (2007). "Breakthrough of the Year: Human Genetic Variation". Science 318 (5858): 1842–1843. doi:10.1126/science.318.5858.1842. PMID 18096770.
- Ramachandran, S; Tang, H; Gutenkunst, RN; Bustamante, CD (2010). "Genetics and Genomics of Human Population Structure". In Speicher, MR; Antonarakis, SE; Motulsky, AG. Vogel and Motulsky’s Human Genetics: Problems and Approaches (4th ed.). Springer. ISBN 3-540-37653-4.
External links
| ||||||
| ||||||||||
| ||||||||||||||||||||||||||||||
| ||||||||||||||||||||||
| ||||||