Hepcidin
| Hepcidin antimicrobial peptide | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
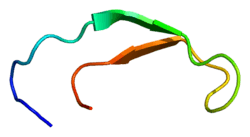 PDB rendering based on 1m4f. | |||||||||||||
| |||||||||||||
| Identifiers | |||||||||||||
| Symbols | HAMP ; HEPC; HFE2B; LEAP1; PLTR | ||||||||||||
| External IDs | OMIM: 606464 HomoloGene: 81623 GeneCards: HAMP Gene | ||||||||||||
| |||||||||||||
| RNA expression pattern | |||||||||||||
 | |||||||||||||
| More reference expression data | |||||||||||||
| Orthologs | |||||||||||||
| Species | Human | Mouse | |||||||||||
| Entrez | 57817 | n/a | |||||||||||
| Ensembl | ENSG00000105697 | n/a | |||||||||||
| UniProt | P81172 | n/a | |||||||||||
| RefSeq (mRNA) | NM_021175 | n/a | |||||||||||
| RefSeq (protein) | NP_066998 | n/a | |||||||||||
| Location (UCSC) | Chr 19: 35.77 – 35.78 Mb | n/a | |||||||||||
| PubMed search | n/a | ||||||||||||
| Hepcidin | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 Solution structure of hepcidin-25.[1] | |||||||||
| Identifiers | |||||||||
| Symbol | Hepcidin | ||||||||
| Pfam | PF06446 | ||||||||
| InterPro | IPR010500 | ||||||||
| SCOP | 1m4f | ||||||||
| SUPERFAMILY | 1m4f | ||||||||
| OPM superfamily | 162 | ||||||||
| OPM protein | 1m4e | ||||||||
| |||||||||
| hepcidin antimicrobial peptide | |
|---|---|
| Identifiers | |
| Symbol | HAMP |
| Entrez | 57817 |
| HUGO | 15598 |
| OMIM | 606464 |
| RefSeq | NM_021175 |
| UniProt | P81172 |
| Other data | |
| Locus | Chr. 19 q13.1 |
Hepcidin is a protein that in humans is encoded by the HAMP gene. Hepcidin is a key regulator of the entry of iron into the circulation in mammals.[2]
In states in which the hepcidin level is abnormally high such as inflammation, serum iron falls due to iron trapping within macrophages and liver cells and decreased gut iron absorption. This typically leads to anemia due to an inadequate amount of serum iron being available for developing red cells. When the hepcidin level is abnormally low such as in hemochromatosis, iron overload occurs due to excessive ferroportin mediated iron influx.
Species
In lab mice hepcidin has been found to have anti-inflammatory properties. This is a negative feedback: it reduces the inflammation which caused the elevated hepcidin level.[3]
Tissues
It is a peptide hormone synthesized mainly in the liver which was discovered in 2000. It reduces dietary iron absorption by reducing iron transport across the gut mucosa (enterocytes); it reduces iron exit from macrophages, the main site of iron storage; and it reduces iron exit from the liver. In all three instances this is accomplished by reducing the transmembrane iron transporter ferroportin.
Structure
Hepcidin exists as a preprohormone (84 amino acids), prohormone (60 amino acids), and hormone (25 amino acids). Twenty- and 22-amino acid metabolites of hepcidin also exist in the urine. Deletion of 5 N-terminal amino acids results in loss of function. The conversion of prohepcidin to hepcidin is mediated by the prohormone convertase furin.[4] This conversion may be regulated by alpha-1 antitrypsin.[5]
Hepcidin is a tightly folded polypeptide with 32% beta sheet character and a hairpin structure stabilized by 4 disulfide bonds. The structure of hepcidin has been determined through solution NMR.[1] NMR studies showed a new model for hepcidin: at ambient temperatures, the protein interconverts between two conformations, which could be individually resolved by temperature variation. The solution structure of hepcidin was determined at 325 K and 253 K in supercooled water. X-ray analysis of a co-crystal with Fab revealed a structure similar to the high-temperature NMR structure.[6]
Function

Hepcidin is a regulator of iron metabolism. Hepcidin inhibits iron transport by binding to the iron export channel ferroportin which is located on the basolateral surface of gut enterocytes and the plasma membrane of reticuloendothelial cells (macrophages). Hepcidin ultimately breaks down the transporter protein in the lysosome. Inhibiting ferroportin prevents iron from being exported and the iron is sequestered in the cells.[7][8] By inhibiting ferroportin, hepcidin prevents enterocytes from allowing iron into the hepatic portal system, thereby reducing dietary iron absorption. The iron release from macrophages is also reduced by ferroportin inhibition. Increased hepcidin activity is partially responsible for reduced iron availability seen in anemia of chronic inflammation. such as renal failure.[9]
Any one of several mutations in hepcidin result in juvenile hemochromatosis. The majority of juvenile hemochromatosis cases are due to mutations in hemojuvelin.[10] Mutations in TMPRSS6 can cause anemia through dysregulation of Hepcidin[11]
Hepcidin has antimicrobial activity against one E. Coli, two staph, one strep, and one fungal species.[12]
Regulation
Hepcidin synthesis and secretion by the liver is controlled by iron stores within macrophages, inflammation, hypoxia, and erythropoiesis. Macrophages communicate with the hepatocyte to regulate hepcidin release into the circulation via eight different proteins: hemojuvelin, heriditrary hemochromatosis protein, transferrin receptor 2, bone morphogenic protein 6 (BMP6), matriptase-2, neogenin, BMP receptors, and transferrin.[13]
History
The peptide was initially named LEAP-1, for Liver-Expressed Antimicrobial Protein.[14] Later, a peptide associated with inflammation was discovered, and named "hepcidin" after it was observed that it was produced in the liver ("hep-") and appeared to have bactericidal properties ("-cide" for "killing").[15] Although it is primarily synthesized in the liver, smaller amounts are synthesised in other tissues such as fat cells.[16]
Hepcidin was first discovered in human urine and serum in 2000.[17]
Soon after this discovery, researchers discovered that hepcidin production in mice increases in conditions of iron overload as well as in inflammation. Genetically modified mice engineered to overexpress hepcidin died shortly after birth with severe iron deficiency, again suggesting a central and not redundant role in iron regulation. The first evidence that linked hepcidin to the clinical condition known as the anemia of inflammation came from the lab of Nancy Andrews in Boston when researchers looked at tissue from two patients with liver tumors with a severe microcytic anemia that did not respond to iron supplements. The tumor tissue appeared to be overproducing hepcidin, and contained large quantities of hepcidin mRNA. Removing the tumors surgically cured the anemia.
Taken together, these discoveries suggested that hepcidin regulates the absorption of iron into the body.
Clinical significance
There are many diseases where failure to adequately absorb iron contributes to iron deficiency and iron deficiency anaemia. The treatment will depend on the hepcidin levels that are present, as oral treatment will be unlikely to be effective if hepcidin is blocking enteral absorption, in which cases parenteral iron treatment would be appropriate. Studies have found that measuring hepcidin would be of benefit to establish optimal treatment,[18] although as this is not widely available, C-reactive protein (CRP) is used as a surrogate marker.
Beta-thalassemia is one of the most common congenital anemias arising from partial or complete lack of β-globin synthesis. Excessive iron absorption is one of the main features of β-thalassemia and can lead to severe morbidity and mortality. The serial analyses of β-thalassemic mice indicate hemoglobin levels decreases over time, while the concentration of iron in the liver, spleen, and kidneys markedly increases. The overload of iron is associated with low levels of hepcidin. It was found that patients with β-thalassemia also have low hepcidin levels. The observations led researchers to hypothesize that more iron is absorbed in β-thalassemia than is required for erythropoiesis and whether increasing the concentration of hepcidin in the body of such patients might be therapeutic, limiting iron overload. It was demonstrated that a moderate increase in expression of hepcidin in β-thalassemic mice limits iron overload, decreases formation of insoluble membrane-bound globins and reactive oxygen species, and improves anemia. Mice with increased hepcidin expression also demonstrated an increase in the lifespan of their red cells, reversal of ineffective erythropoiesis and splenomegaly, and an increase in total hemoglobin levels. This data suggests that therapeutics that would increase hepcidin levels or act as hepcidin agonists might help treat the abnormal iron absorption in individuals with β-thalassemia and related disorders.[19][20] Bolus doses of vitamin D2 (100,000iu) decrease circulating hepcidin rapidly, thereby enhancing or increasing iron levels in the bloodstream. Circulating hepcidin began to decline 2 hours after a bolus of vitamin D2 before circulating 25(OH)D begins to rise. Optimal function of hepcidin would seem to be predicated upon adequate presence of vitamin D in the blood.[21][22]
References
- ↑ 1.0 1.1 PDB 1M4F; Hunter HN, Fulton DB, Ganz T, Vogel HJ (October 2002). "The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis". J. Biol. Chem. 277 (40): 37597–603. doi:10.1074/jbc.M205305200. PMID 12138110.
- ↑ Ganz T (August 2003). "Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation". Blood 102 (3): 783–8. doi:10.1182/blood-2003-03-0672. PMID 12663437.
- ↑ De Domenico I, Zhang TY, Koening CL, Branch RW, London N, Lo E, Daynes RA, Kushner JP, Li D, Ward DM, Kaplan J (July 2010). "Hepcidin mediates transcriptional changes that modulate acute cytokine-induced inflammatory responses in mice". J. Clin. Invest. 120 (7): 2395–405. doi:10.1172/JCI42011. PMC 2898601. PMID 20530874.
- ↑ Valore EV, Ganz T (2008). "Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin". Blood Cells Mol. Dis. 40 (1): 132–8. doi:10.1016/j.bcmd.2007.07.009. PMC 2211381. PMID 17905609.
- ↑ Pandur E, Nagy J, Poór VS, Sarnyai A, Huszár A, Miseta A, Sipos K (April 2009). "Alpha-1 antitrypsin binds preprohepcidin intracellularly and prohepcidin in the serum". FEBS J. 276 (7): 2012–21. doi:10.1111/j.1742-4658.2009.06937.x. PMID 19292870.
- ↑ PDB 3H0T; Jordan JB, Poppe L, Haniu M, Arvedson T, Syed R, Li V, Kohno H, Kim H, Schnier PD, Harvey TS, Miranda LP, Cheetham J, Sasu BJ (September 2009). "Hepcidin revisited, disulfide connectivity, dynamics, and structure". J. Biol. Chem. 284 (36): 24155–67. doi:10.1074/jbc.M109.017764. PMC 2782009. PMID 19553669.
- ↑ Rossi E (August 2005). "Hepcidin--the iron regulatory hormone". Clin Biochem Rev 26 (3): 47–9. PMC 1240030. PMID 16450011.
- ↑ "Mechanistic and regulatory aspects of intestinal iron absorption.". AJP: Gastrointestinal and Liver Physiology 307: G397–409. Aug 2014. doi:10.1152/ajpgi.00348.2013. PMID 24994858.
- ↑ Ashby DR, Gale DP, Busbridge M, Murphy KG, Duncan ND, Cairns TD, Taube DH, Bloom SR, Tam FW, Chapman RS, Maxwell PH, Choi P (May 2009). "Plasma hepcidin levels are elevated but responsive to erythropoietin therapy in renal disease". Kidney Int. 75 (9): 976–81. doi:10.1038/ki.2009.21. PMID 19212416.
- ↑ Core, AB; Canali, S; Babitt, JL (2014). "Hemojuvelin and bone morphogenetic protein (BMP) signaling in iron homeostasis.". Frontiers in pharmacology 5: 104. doi:10.3389/fphar.2014.00104. PMID 24860505.
- ↑ Iron-Deficiency Anemia: New Insights for the Healthcare Professional: 2011 Edition. Scholarly Media LLC. Dec 2012. ISBN 978-1-4649-8960-5.
- ↑ http://www.uniprot.org/uniprot/P81172
- ↑ Zhao, J, JCI (2013);123(6):2337doi:10.1172/JCI67225
- ↑ Krause A, Neitz S, Mägert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K (September 2000). "LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity". FEBS Lett. 480 (2–3): 147–50. doi:10.1016/S0014-5793(00)01920-7. PMID 11034317.
- ↑ Park CH, Valore EV, Waring AJ, Ganz T (March 2001). "Hepcidin, a urinary antimicrobial peptide synthesized in the liver". J. Biol. Chem. 276 (11): 7806–10. doi:10.1074/jbc.M008922200. PMID 11113131.
- ↑ Bekri S, Gual P, Anty R, Luciani N, Dahman M, Ramesh B, Iannelli A, Staccini-Myx A, Casanova D, Ben Amor I, Saint-Paul MC, Huet PM, Sadoul JL, Gugenheim J, Srai SK, Tran A, Le Marchand-Brustel Y (September 2006). "Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and NASH". Gastroenterology 131 (3): 788–96. doi:10.1053/j.gastro.2006.07.007. PMID 16952548.
- ↑ Kemna EH, Tjalsma H, Willems HL, Swinkels DW (January 2008). "Hepcidin: from discovery to differential diagnosis". Haematologica 93 (1): 90–7. doi:10.3324/haematol.11705. PMID 18166790.
- ↑ Bregman DB, Morris D, Koch TA, He A, Goodnough LT (February 2013). "Hepcidin levels predict nonresponsiveness to oral iron therapy in patients with iron deficiency anemia". Am. J. Hematol. 88 (2): 97–101. doi:10.1002/ajh.23354. PMID 23335357.
- ↑ Gardenghi S, Ramos P, Marongiu MF, Melchiori L, Breda L, Guy E, Muirhead K, Rao N, Roy CN, Andrews NC, Nemeth E, Follenzi A, An X, Mohandas N, Ginzburg Y, Rachmilewitz EA, Giardina PJ, Grady RW, Rivella S (December 2010). "Hepcidin as a therapeutic tool to limit iron overload and improve anemia in β-thalassemic mice". J. Clin. Invest. 120 (12): 4466–77. doi:10.1172/JCI41717. PMC 2993583. PMID 21099112.
- ↑ Kroot JJ, Tjalsma H, Fleming RE, Swinkels DW (December 2011). "Hepcidin in human iron disorders: diagnostic implications". Clin. Chem. 57 (12): 1650–69. doi:10.1373/clinchem.2009.140053. PMID 21989113.
- ↑ M. Hewison, B.W. HOllis, unpublished data, 2013
- ↑ Hollis, BW; Wagner, CL (Dec 2013). "Clinical review: The role of the parent compound vitamin D with respect to metabolism and function: Why clinical dose intervals can affect clinical outcomes.". The Journal of Clinical Endocrinology and Metabolism 98 (12): 4619–28. doi:10.1210/jc.2013-2653. PMID 24106283.
Further reading
- Camaschella C (2005). "Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders". Blood 106 (12): 3710–7. doi:10.1182/blood-2005-05-1857. PMID 16030190.
External links
- hepcidin at the US National Library of Medicine Medical Subject Headings (MeSH)
- Intrinsic LifeSciences - Hepcidin Research Facility, The BioIron Company
- Hepcidinanalysis.com - Service for Hepcidin measurements: Scientific Research, Patients and Clinical Trials
- Protein Data Bank Page
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||