Haplotype estimation
In genetics, haplotype estimation (also known as, phasing) refers to the process of statistical estimation of haplotypes from genotype data. The most common situation arises when genotypes are collected at a set of polymorphic sites from a group of individuals. For example, in human genetics genome-wide association studies collect genotypes in thousands of individuals at between 200,000-5,000,000 SNPs using microarrays. Haplotype estimation methods are used in the analysis of these datasets and allow genotype imputation [1][2] of alleles from reference databases such as the HapMap Project and the 1000 Genomes Project. Haplotype estimation is sometimes referred to as phasing.
Genotypes and haplotypes

Genotypes measure the unordered combination of alleles at each site, whereas haplotypes are the two sequence of alleles that have been inherited together from the individuals parents. When there are 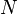 heterozygous genotypes present an individuals set of genotypes there will be
heterozygous genotypes present an individuals set of genotypes there will be 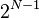 possible pairs of haplotypes that could underlie the genotypes. If there are missing genotypes then the number of possible haplotype pairs increases.
possible pairs of haplotypes that could underlie the genotypes. If there are missing genotypes then the number of possible haplotype pairs increases.
Haplotype estimation methods
There is a large literature of statistical methods that have been proposed for estimation of haplotypes. Some of the earliest approaches used a simple multinomial model in which each possible haplotype consistent with the sample was given an unknown frequency parameter and these parameters were estimated with an EM algorithm. These approaches were only able to handle small numbers of sites at once, although sequential versions were later developed, specifically the SNPHAP method.
The most accurate and widely used methods for haplotype estimation utilize some form of hidden Markov model (HMM) to carry out inference. For a long time the method PHASE[3] was the most accurate method. PHASE was the first method to utilize ideas from coalescent theory concerning the joint distribution of haplotypes. This method used a Gibbs sampling approach in which each individuals haplotypes were updated conditional upon the current estimates of haplotypes from all other samples. Approximations to the distribution of a haplotype conditional upon a set of other haplotypes were used for the conditional distributions of the Gibbs sampler. PHASE was used to estimate the haplotypes from the HapMap Project. PHASE was limited by its speed and was not applicable to datasets from genome-wide association studies.
The fastPHASE [4] and BEAGLE methods [5] introduced haplotype cluster models applicable to GWAS-sized datasets. Subsequently the IMPUTE2[6] and MaCH[7] methods were introduced that were similar to the PHASE approach but much faster. These methods iteratively update the haplotype estimates of each sample conditional upon a subset of K haplotype estimates of other samples. IMPUTE2 introduced the idea of carefully choosing which subset of haplotypes to condition on to improve accuracy. Accuracy increases with K but with quadratic 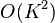 computational complexity.
computational complexity.
The SHAPEIT1 method made a major advance by introducing a linear 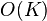 complexity method that operates only on the space of haplotypes consistent with an individual’s genotypes.[8] The HAPI-UR method subsequently proposed a very similar method.[9] SHAPEIT2 [10] combines the best features of SHAPEIT1 and IMPUTE2 to improve efficiency and accuracy.
complexity method that operates only on the space of haplotypes consistent with an individual’s genotypes.[8] The HAPI-UR method subsequently proposed a very similar method.[9] SHAPEIT2 [10] combines the best features of SHAPEIT1 and IMPUTE2 to improve efficiency and accuracy.
Software
See also
- imputation: predict missing genotypes using known haplotypes
References
- ↑ Marchini, J.; Howie, B. (2010). "Genotype imputation for genome-wide association studies". Nature Reviews Genetics 11 (7): 499–511. doi:10.1038/nrg2796. PMID 20517342.
- ↑ Howie, B.; Fuchsberger, C.; Stephens, M.; Marchini, J.; Abecasis, G. A. R. (2012). "Fast and accurate genotype imputation in genome-wide association studies through pre-phasing". Nature Genetics 44 (8): 955–959. doi:10.1038/ng.2354. PMID 22820512.
- ↑ Stephens, M.; Smith, N. J.; Donnelly, P. (2001). "A New Statistical Method for Haplotype Reconstruction from Population Data". The American Journal of Human Genetics 68 (4): 978–989. doi:10.1086/319501. PMC 1275651. PMID 11254454.
- ↑ Scheet, P.; Stephens, M. (2006). "A Fast and Flexible Statistical Model for Large-Scale Population Genotype Data: Applications to Inferring Missing Genotypes and Haplotypic Phase". The American Journal of Human Genetics 78 (4): 629–644. doi:10.1086/502802. PMC 1424677. PMID 16532393.
- ↑ Browning, S. R.; Browning, B. L. (2007). "Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies by Use of Localized Haplotype Clustering". The American Journal of Human Genetics 81 (5): 1084–1097. doi:10.1086/521987. PMC 2265661. PMID 17924348.
- ↑ Howie, B. N.; Donnelly, P.; Marchini, J. (2009). Schork, Nicholas J, ed. "A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies". PLoS Genetics 5 (6): e1000529. doi:10.1371/journal.pgen.1000529. PMC 2689936. PMID 19543373.
- ↑ Li, Y.; Willer, C. J.; Ding, J.; Scheet, P.; Abecasis, G. A. R. (2010). "MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes". Genetic Epidemiology 34 (8): 816–834. doi:10.1002/gepi.20533. PMC 3175618. PMID 21058334.
- ↑ Delaneau, O.; Marchini, J.; Zagury, J. F. O. (2011). "A linear complexity phasing method for thousands of genomes". Nature Methods 9 (2): 179–181. doi:10.1038/nmeth.1785. PMID 22138821.
- ↑ Williams, A. L.; Patterson, N.; Glessner, J.; Hakonarson, H.; Reich, D. (2012). "Phasing of Many Thousands of Genotyped Samples". The American Journal of Human Genetics 91 (2): 238–251. doi:10.1016/j.ajhg.2012.06.013. PMC 3415548. PMID 22883141.
- ↑ Delaneau, O.; Zagury, J. F.; Marchini, J. (2012). "Improved whole-chromosome phasing for disease and population genetic studies". Nature Methods 10 (1): 5–6. doi:10.1038/nmeth.2307. PMID 23269371.