Gap penalty
The gap penalty is a scoring system used in bioinformatics for aligning a small portion of genetic code, more accurately, fragmented genetic sequence, also termed, reads against a reference genetic sequence (e.g. The Human Genome). The biological process of protein synthesis namely, transcription and translation or DNA replication can produce errors resulting in mutations in the final nucleic acid sequence. Therefore, in order to make more accurate decisions in aligning reads, mutations are annotated as gaps in the sequence. Gaps are penalised via various Gap Penalty scoring methods. Gaps in a DNA sequence refer to substitutions or indels in a sequence, where indels can be insertions or deletions in the sequence. Insertions or deletions occur due to single mutations, unbalanced crossover in meiosis, slipped strand mispairing in the replication process and chromosomal translocation.[1] In alignments gaps are represented as contiguous dashes on a protein/DNA sequence alignment.[2] The scoring that occurs in Gap Penalty allows for the optimisation of sequence alignment in order to obtain the best alignment possible based on the information available. The three main types of gap penalties are constant, linear and affine gap penalty.[3]
The notion of a gap in an alignment is important in many biological applications, since the insertions or deletions comprise an entire sub-sequence and often occur from a single mutational event.[4] Furthermore, single mutational events can create gaps of different sizes. Therefore, when scoring, the gaps need to be scored as a whole when aligning two sequences of DNA. Considering multiple gaps in a sequence as a larger single gap will reduce the assignment of a high cost to the mutations. For instance, two protein sequences may be relatively similar however, may differ at certain intervals as one protein may have a different subunit compared to the other. Representing these differing sub-sequences as gaps will allow us to treat these cases as “good matches” even though there are long consecutive runs with indel operations in the sequence. Therefore, using a good gap penalty model will avoid low scores in alignments and improve the chances of finding a true alignment.[4]
Gap Penalty applications can be applied outside biological cases. For instance, gap penalty is used in the diff function in Unix to compute the minimal difference between two files. Other applications include spell checking, plagiarism detection and speech recognition in software algorithms to name a few.
Types
Constant
This is the simplest type of gap penalty: a fixed negative score is given to every gap, regardless of the its length.[4][5]
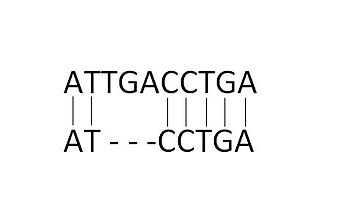
Linear
Compared to the constant gap penalty, the linear gap penalty takes into account the length (L) of each insertion/deletion in the gap. Therefore, if the penalty for each inserted/deleted element is B and the length of the gap L; the total gap penalty would be the product of the two BL.[6] This method favors shorter gaps, with total score decreasing with each additional gap.
Affine
The most widely used gap penalty function is the affine gap penalty. The affine gap penalty combines the components in both the constant and linear gap penalty, taking the form A+(B⋅L). This introduces new terms, A is known as the gap opening penalty, B the gap extension penalty and L the length of the gap. Gap opening refers to the cost required to open a gap of any length, and gap extension the cost to extend the length of an existing gap by 1.[7] Often it is unclear as to what the values A and B should be as it differs according to purpose. In general, if the interest is to find closely related matches (e.g. removal of vector sequence during genome sequencing), a higher gap penalty should be used to reduce gap openings. On the other hand, gap penalty should be lowered when interested in finding a more distant match.[6] The relationship between A and B also have an effect on gap size. If the size of the gap was important, a small A and large B (more costly to extend gap) is used and vice versa.
Convex
Using the affine gap penalty requires the assigning of fixed penalty values for both opening and extending a gap. This can be too rigid for use in a biological context.[8]
The logarithmic gap takes the form G(L) = A + ClnL and was proposed as studies had shown the distribution of indel sizes obey a power law.[9] Another proposed issue with the use of affine gaps is the favoritism of aligning sequences with shorter gaps. Logarithmic gap penalty was invented to modify the affine gap so that long gaps are desirable.[8] However, in contrast to this, it has been found that using logarithmatic models had produced poor alignments when compared to affine models.[9]
Profile-based variable gap penalties
Profile–profile alignment algorithms are powerful tools for detecting protein homology relationships with improved alignment accuracy.[10] Profile-profile alignments are based on the statistical indel frequency profiles from multiple sequence alignments generated by PSI-BLAST searches.[10] Rather than using substitution matrices to measure the similarity of amino acid pairs, profile–profile alignment methods require a profile-based scoring function to measure the similarity of profile vector pairs.[10] Profile-profile alignments employ gap penalty functions. The gap information is usually used in the form of indel frequency profiles, which is more specific for the sequences to be aligned. ClustalW and MAFFT adopted this kind of gap penalty determination for their multiple sequence alignments.[10] Alignment accuracies can be improved using this model, especially for proteins with low sequence identity. Some profile–profile alignment algorithms also run the secondary structure information as one term in their scoring functions, which improves alignment accuracy.[10]
Applications
Global alignment
A global alignment performs an end-to-end alignment of the query sequence with the reference sequence. Ideally, this alignment technique is most suitable for closely related sequences of similar lengths. The Needleman-Wunsch algorithm is a dynamic programming technique used to conduct global alignment. Essentially, the algorithm divides the problem into a set of sub-problems, than uses the results of the sub-problems to reconstruct a solution to the original query.[11]
General steps to perform a global alignment:[12]
- Create a scoring matrix
- Fill in the scoring matrix - the matrix is filled with the maximum score possible starting in the top left corner and subsequently filling in the neighboring cells (left, right and diagonal).
- Trace back - trace back starting from the lowest right hand cell and choosing the minimal score trace to find the best alignment.
Pseudocode
procedure Needleman-Wunsch Algorithm S[i,j] = min { S[i-1, j-1] if match S[i-1, j-1] + 1 if mismatch S[i-1, j] + 1 S[i, j-1] + 1 } end procedure
Semi-global alignment
The use of semi-global alignment exists to find a particular match within a large sequence. An example includes seeking promoters within a DNA sequence. Unlike global alignment, it compromises of no end gaps in one or both sequences. If the end gaps are penalized in one sequence 1 but not in sequence 2, it produces an alignment that contains sequence 1 within sequence 2.
Local alignment

A local sequence alignment matches a contiguous sub-section of one sequence with a contiguous sub-section of another.[13] The Smith-Waterman algorithm is motivated by giving scores for matches and mismatches. Matches increase the overall score of an alignment whereas mismatches decrease the score. A good alignment then has a positive score and a poor alignment has a negative score. The local algorithm finds an alignment with the highest score by considering only alignments that score positives and picking the best one from those. The algorithm is a Dynamic programming algorithm. When comparing proteins, one uses a similarity matrix which assigns a score to each possible residue. The score should be positive for similar residues and negative for dissimilar residues pair. Gaps are usually penalized using a linear gap function that assigns an initial penalty for a gap opening, and an additional penalty for gap extensions, increasing the gap length.
Scoring matrix

Substitution matrices such as BLOSUM are used for sequence alignment of proteins.[14] A Substitution matrix assigns a score for aligning any possible pair of residues.[14] In general, different substitution matrices are tailored to detecting similarities among sequences that are diverged by differing degrees. A single matrix may be reasonably efficient over a relatively broad range of evolutionary change.[14] The BLOSUM-62 matrix is one of the best substitution matrices for detecting weak protein similarities.[14] BLOSUM matrices with high numbers are designed for comparing closely related sequences, while those with low numbers are designed for comparing distant related sequences. For example, BLOSUM-80 is used for alignments that are more similar in sequence, and BLOSUM-45 is used for alignments that have diverged from each other.[14] For particularly long and weak alignments, the BLOSUM-45 matrix may provide the best results. Short alignments are more easily detected using a matrix with a higher "relative entropy" than that of BLOSUM-62. The BLOSUM series does not include any matrices with relative entropies suitable for the shortest queries.[14]
Indels
During DNA Replication, the replication machinery is prone to making two types of errors while duplicating the DNA. These two replication errors are insertions and deletions of single DNA bases from the DNA strand (indels).[15] Indels can have severe biological consequences by causing mutations in the DNA strand that could result in the inactivation or over activation of the target protein. For example if a one or two nucleotide indel occurs in a coding sequence the result will be a shift in the reading frame, or a frameshift mutation that may render the protein inactive.[15] The biological consequences of indels are often deleterious and are frequently associated with human pathologies such as cancer. However not all indels are frameshift mutations. If indels occur in trinucleotides, the result is an extension of the protein sequence that may also have implications on protein function.[15]
Comparing time complexities
The use of alignment in computational biology often involves sequences of varying lengths. It is important to pick a model that would efficiently run at a known input size. The time taken to run the algorithm is known as the time complexity.
| Type | Time |
|---|---|
| Constant gap penalty | O(mn) |
| Affine gap penalty | O(mn) |
| Convex gap penalty | O(mn lg(m+n)) |
Assigning Gap Penalty Values
Gap penalty values are designed to reduce the score when an alignment has been disturbed by indels. The value should be small enough to allow a previously accumulated alignment to continue with an insertion in one of the sequences but should not be so large that this previous alignment score is removed completely. There are two strategies when assigning values to gaps:
- Keep the score similar regardless of gap length. Allow a constant overall gap penalty regardless of gap length. Therefore assign no gap extension penalty and only penalize the sequence when there is a gap open. This will penalize a large gap by the same extent as a small gap.[16]
- Make the score becomes larger as a linear function of gap length. Have a larger gap opening penalty followed by a gap extension penalty that is smaller than the gap open penalty. This will penalize several small gaps by the same extent as 1 large gap.[16]
Challenges
There are a few challenges when it comes to working with gaps. When working with popular algorithms there seems to be little theoretical basis for the form of the gap penalty functions.[17] Consequently, for any alignment situation gap placement must be empirically determined.[17] Also, pairwise alignment gap penalties,such as the affine gap penalty, are often implemented independent of the amino acid types in the inserted or deleted fragment or at the broken ends, despite evidence that specific residue types are preferred in gap regions.[17] Finally, alignment of sequences implies alignment of the corresponding structures, but the relationships between structural features of gaps in proteins and their corresponding sequences are only imperfectly known. Because of this incorporating structural information into gap penalties is difficult to do.[17] Some algorithms use predicted or actual structural information to bias the placement of gaps. However, only a minority of sequences have known structures, and most alignment problems involve sequences of unknown secondary and tertiary structure.[17]
References
- ↑ Carroll, Ridge, Clement, Snell, Hyrum , Perry, Mark, Quinn (January 1, 2007). "Effects of Gap Open and Gap Extension Penalties". International Journal Of Bioinformatics Research And Applications. Retrieved 09/09/14. Check date values in:
|accessdate=(help) - ↑ "Glossary". Rosalind. Rosalind Team. Retrieved 11/09/14. Check date values in:
|accessdate=(help) - ↑ "Glossary". Rosalind. Rosalind Team. Retrieved 11/09/14. Check date values in:
|accessdate=(help) - ↑ 4.0 4.1 4.2 "Algorithms for Molecular Biology". BioMed Central. 2006-01-01. Retrieved 13/09/14. Check date values in:
|accessdate=(help) - ↑ "Glossary - Constant Gap Penalty". Rosalind. Rosalind Team. 12 Aug 2014. Retrieved 12 Aug 2014.
- ↑ 6.0 6.1 Hodgman C, French A, Westhead D, (2009). BIOS Instant Notes in Bioinformatics. Garland Science. pp. 143–144. ISBN 0203967240.
- ↑ "Global Alignment with Scoring Matrix and Affine Gap Penalty". Rosalind. Rosalind Team. 2/7/2012. Retrieved 2014-09-12. Check date values in:
|date=(help) - ↑ 8.0 8.1 Sung, Wing-Kin (2011). Algorithms in Bioinformatics : A Practical Introduction. CRC Press. pp. 42–47. ISBN 1420070347.
- ↑ 9.0 9.1 Cartwright, Reed (5/12/2006). "Logarithmic gap costs decrease alignment accuracy". BMC Bioinformatics. doi:10.1186/1471-2105-7-527. Retrieved 2014-09-10. Check date values in:
|date=(help) - ↑ 10.0 10.1 10.2 10.3 10.4 Wang C, Yan RX, Wang XF, Si JN, Zhang Z (12 October 2011). "Comparison of linear gap penalties and profile-based variable gap penalties in profile-profile alignments". Comput Biol Chem 35 (5): 308–318. doi:10.1016/j.compbiolchem.2011.07.006. PMID 22000802.
- ↑ Lesk, Arthur M (2013-07-26). "bioinformatics". Encyclopedia Britannica. Encyclopedia Britannica. Retrieved 2014-09-12.
- ↑ "Global alignment of two sequences - Needleman-Wunsch Algorithm". Value @ Amrita: Virtual Amrita Laboratories Universalizing Education. Amrita Vishwa Vidyapeetham University. Retrieved 12/09/14. Check date values in:
|accessdate=(help) - ↑ Vingron, M.; Waterman, M. S. (1994). "Sequence alignment and penalty choice. Review of concepts, case studies and implications". Journal of molecular biology 235 (1): 1–12. doi:10.1016/S0022-2836(05)80006-3. PMID 8289235.
- ↑ 14.0 14.1 14.2 14.3 14.4 14.5 "BLAST substitution matrices". NCBI. Retrieved 2012-11-27.
- ↑ 15.0 15.1 15.2 Garcia-Diaz, Miguel (2006). "Mechanism of a genetic glissando: structural biology of indel mutations". Trends in Biochemical Sciences 31 (4). doi:10.1016/j.tibs.2006.02.004.
- ↑ 16.0 16.1 "About Gaps In Sequence Alignments". EMBL-EBI. Retrieved 2012-11-27.
- ↑ 17.0 17.1 17.2 17.3 17.4 Wrabl JO, Grishin NV (1 January 2004). "Gaps in structurally similar proteins: towards improvement of multiple sequence alignment". Proteins 54 (1): 71–87. doi:10.1002/prot.10508. PMID 14705025.
Further reading
- Taylor WR, Munro RE (1997). "Multiple sequence threading: conditional gap placement". Fold Des 2 (4): S33–9. doi:10.1016/S1359-0278(97)00061-8.
- Taylor WR (1996). "A non-local gap-penalty for profile alignment". Bull Math Biol 58 (1): 1–18. doi:10.1007/BF02458279. PMID 8819751.
- Vingron M, Waterman MS (1994). "Sequence alignment and penalty choice. Review of concepts, case studies and implications". J Mol Biol 235 (1): 1–12. doi:10.1016/S0022-2836(05)80006-3. PMID 8289235.
- Panjukov VV (1993). "Finding steady alignments: similarity and distance". Comput Appl Biosci 9 (3): 285–90. doi:10.1093/bioinformatics/9.3.285. PMID 8324629.
- Alexandrov NN (1992). "Local multiple alignment by consensus matrix". Comput Appl Biosci 8 (4): 339–45. doi:10.1093/bioinformatics/8.4.339. PMID 1498689.
- Hein J (1989). "A new method that simultaneously aligns and reconstructs ancestral sequences for any number of homologous sequences, when the phylogeny is given". Mol Biol Evol 6 (6): 649–68. PMID 2488477.
- Henneke CM (1989). "A multiple sequence alignment algorithm for homologous proteins using secondary structure information and optionally keying alignments to functionally important sites". Comput Appl Biosci 5 (2): 141–50. doi:10.1093/bioinformatics/5.2.141. PMID 2751764.
- Reich JG, Drabsch H, Daumler A (1984). "On the statistical assessment of similarities in DNA sequences". Nucleic Acids Res 12 (13): 5529–43. doi:10.1093/nar/12.13.5529. PMC 318937. PMID 6462914.