Fibrodysplasia ossificans progressiva
| Fop Disease | |
|---|---|
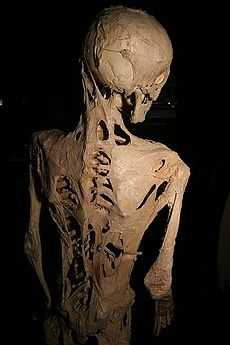 The effects of Fibrodysplasia ossificans progressiva, a disease which causes damaged soft tissue to regrow as bone. Sufferers are slowly imprisoned by their own skeletons. | |
| Classification and external resources | |
| ICD-10 | M61.1 |
| ICD-9 | 728.11 |
| OMIM | 135100 |
| DiseasesDB | 8732 |
| eMedicine | derm/609 |
| MeSH | D009221 |
Fibrodysplasia ossificans progressiva (FOP), sometimes referred to as Stone Man Syndrome, is an extremely rare disease of the connective tissue. A mutation of the body's repair mechanism causes fibrous tissue (including muscle, tendon, and ligament) to be ossified spontaneously or when damaged. In many cases, injuries can cause joints to become permanently frozen in place. Surgical removal of the extra bone growths has been shown to cause the body to "repair" the affected area with more bone.[1]
Signs and symptoms
For unknown reasons, children born with FOP have deformed big toes, possibly missing a joint or simply presenting with a notable lump at the minor joint. The first "flare-up" that leads to the formation of FOP bones usually occurs before the age of 10. The bone growth progresses from the top downward, just as bones grow in fetuses. A child with FOP will typically develop bones starting at the neck, then on the shoulders, arms, chest area and finally on the feet.
Specifically, FOP involvement is typically seen first in the dorsal, axial, cranial and proximal regions of the body. Later the disease progresses in the ventral, appendicular, caudal and distal regions of the body.[2] However, it does not necessarily occur in this order due to injury-caused flare-ups. Often, the tumor-like lumps that characterize the disease appear suddenly. This condition causes loss of mobility to affected joints, including inability to fully open the mouth limiting speech and eating. Extra bone formation around the rib cage restricts the expansion of lungs and diaphragm causing breathing complications.
Since the disease is so rare, the symptoms are often misdiagnosed as cancer or fibrosis. This leads physicians to order biopsies, which can actually exacerbate the growth of these lumps. However, those born with FOP tend to have malformed toes or thumbs which help distinguish this disorder from other skeletal problems.[3]
Causes
FOP is caused by an autosomal dominant allele on chromosome 2q23-24.[4] The allele has variable expressivity, but complete penetrance. Most cases are caused by spontaneous mutation in the gametes; most people with FOP cannot or choose not to have children. A similar but less catastrophic disease is fibrous dysplasia, which is caused by a post-zygotic mutation.
A mutation in the gene ACVR1 (also known as activin-like kinase 2 [ALK-2]) is responsible for the disease.[5] ACVR1 encodes activin receptor type-1, a BMP type-1 receptor. The mutation causes substitution of codon 206 from arginine to histidine in the ACVR1 protein.[6] This substitution causes abnormal activation of ACVR1, leading to the transformation of connective tissue and muscle tissue into a secondary skeleton. This causes endothelial cells to transform to mesenchymal stem cells and then to bone.[7]
Genetics
FOP or Stone Man Syndrome is an autosomal dominant disorder that affects individuals who are heterozygous with a homozygous recessive partner, therefore their children will have 50% chance of being affected. Two affected individuals can produce unaffected children. The phenotypes of those who are homozygous dominant have more severe effects compared to those with heterozygous phenotype.[8]
The gene that causes ossification is normally deactivated after a fetus's bones are formed in the womb, but in patients with FOP, the gene keeps working. Aberrant bone formation in patients with FOP occurs when injured connective tissue or muscle cells at the sites of injury or growth incorrectly express an enzyme for bone repair during apoptosis (self-regulated cell death), resulting in lymphocytes containing excess bone morphogenetic protein 4 (BMP4) provided during the immune system response. The bone that results occurs independently of the normal skeleton, forming its own discrete skeletal elements. These elements, however, can fuse with normal skeletal bone.[9] Interestingly, the diaphragm, tongue, and extra-ocular muscles are spared in this process, as well as cardiac and smooth muscle.[2] Since the incorrect enzyme remains unresolved within the immune response, the body continues providing the incorrect BMP4-containing lymphocytes. BMP4 is a product that contributes to the development of the skeleton in the normal embryo.[10]

DNA sequencing electropherograms of a typical FOP patient can differ when being compared to two other patients. The cause of this mutation is in the ACVR1 gene. This gene provides instruction for a protein known as morphogenetic protein (BMP). This protein is responsible for growth and development of bone and muscles. Scientists and researchers theorize that a mutation in the ACVR1 changes the shape of the receptor and disrupts certain mechanisms that control the receptor's activity. There is a certain molecule, otherwise known as ligands, that binds at the site to cause this reaction to activate with which it forms a complex. Due to the mutation, however, the bind site is modified and no longer stops the reaction.[11] The end result is an overgrowth of bone and cartilage and fusion of joints.[12]
This type of genetic disorder is so rare that only 1 in 2 million people worldwide acquire it. As it is such a rare disorder, only a few are reported at all. Most of the cases of FOP were results of a new gene mutation: these people had no history of this particular disorder in their family. There are some cases which have shown people inheriting the mutation from one affected parent.[13]
Diagnosis
Outbreaks may be measurable clinically by elevated levels of alkaline phosphatase and bone-specific alkaline phosphatase.[14]
Treatment
There is no known cure for FOP. Attempts to surgically remove the bone result in more robust bone growth.[15] While under anesthesia, patients with FOP may face problems, which include difficulties with intubation, restrictive pulmonary disease, and changes in the electrical conduction system of the heart.[16] Activities that increase the risk of falling should be avoided, as injuries from falling can provoke the growth of bone.[1]
In 1999, scientists discovered that squalamine in sharks[17] might be useful in treating those suffering from FOP.[18] Squalamine is antiangiogenic and can prevent the growth of blood vessels in cartilaginous tissue, thus preventing creation of bone in sharks. The Genaera Corporation announced a trial of squalamine in 2002[19][20] but terminated about 2007. (Note that squalene is a different compound, also found in sharks, that has no such properties.) Clinical trials of isotretinoin, etidronate with oral corticosteroids, and perhexiline maleate have failed to demonstrate effectiveness, though the variable course of the disease and small numbers of patients leave some room for uncertainty.[14] In April 2013 the La Jolla Pharmaceutical Company was granted orphan drug status for testing of 4-(6-(4-(piperazin-1-yl)phenyl_pyrazolo[1,5-a]pyrimidin-3-yl)quinoline hydrochloride for treatment of FOP.[21]
Researchers believe that specific kinase inhibitors can be developed that will block the aberrant ACVR1 activity, and are actively investigating dorsomorphin and K02288 as lead compounds with the intention of developing effective therapies.[22][23] For example, the more potent dorsomorphin derivative LDN-193189 reduced ossification in a transgenic mouse model, in which the engineering of adult ACVR1 activity created an inflammation-dependent ossification sensitive to corticosteroid treatment.[24]
Another major direction in research is the development of therapeutics based on allele-specific RNA interference to block the aberrant gene from directing production of ACVR1. However, effective treatment by this means may require a better knowledge of what cell types are responsible for the disease, so that inhibitory RNA can be produced from them in the long term.[25]
Although this disorder is currently incurable, understanding and researching the cause of bone formation in FOP could aid in the treatment of other bone disorders, especially common ones such as fractures, hip replacement surgery, and other heterotopic ossifications that occur in trauma or burn victims.[26]
Epidemiology
A study has determined that it affects approximately 1 in every 2 million people.[27]
Cases
Since the 1800s, there have been references in medicine describing people who apparently "turned to stone"; some of these cases may be attributable to FOP.
The best known FOP case is that of Harry Eastlack (1933–1973). His condition began to develop at the age of ten, and by the time of his death from pneumonia in November 1973, six days before his 40th birthday, his body had completely ossified, leaving him able to move only his lips.
Shortly before Eastlack's death, he made it known that he wanted to donate his body to science, in the hope that in death, he would be able to help find a cure for this little-understood and particularly cruel disease. Pursuant to his wishes, his preserved skeleton is now kept at the Mütter Museum in Philadelphia, and has proven to be an invaluable source of information in the study of FOP.
There have approximately been 700 confirmed cases across the globe from an estimated 2500.[28]
See also
External links
- ClinicalTrials.gov search interface
- Clementia's phase 2 clinical trial enrolment
References
- ↑ 1.0 1.1 Clinical Reviews in Bone and Mineral Metabolism
- ↑ 2.0 2.1 Fibrodysplasia ossificans progressiva. Frederick S. Kaplan, MD, Martine Le Merrer, MD, PhD, Professor of Genetics, David L. Glaser, MD, Robert J. Pignolo, MD, PhD, Robert Goldsby, MD, Joseph A. Kitterman, MD, Jay Groppe, PhD, and Eileen M. Shore, PhD
- ↑ "Fibrodysplasia ossificans progressiva". Lister Hill National Center for Biomedical Communications. Retrieved 2013-12-12.
- ↑ Feldman, G. "A recurrent mutation in the BMP type I receptor ACVR1 ( HSC'13) causes inherited and sporadic fibrodysplasia ossificans progressiva." (PDF).
- ↑ Shore EM; Xu M; Feldman GJ et al. (2006). "A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva". Nat. Genet. 38 (5): 525–527. doi:10.1038/ng1783. PMID 16642017.
- ↑ News Release of FOP's Cause
- ↑ Dinther et al. (2010). "ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation". Journal of Bone and Mineral Research: 091211115834058–35. doi:10.1359/jbmr.091110.
- ↑ Cummings, Michael R. "Human Heredity: Principles and Issues". Cengage Learning, 2011, 2009. p.77
- ↑ Insights from a Rare Genetic Disorder of Extra-Skeletal Bone Formation, Fibrodysplasia Ossificans Progressiva (FOP). Eileen M. Shore and Frederick S. Kaplan
- ↑ Kierszenbaum, Abraham (2002). Histology and cell biology. New York: Mosby. ISBN 978-0-323-01639-1.
- ↑ "ACR1", "Genetics Home Reference", February 18, 2014.
- ↑ "Fibrodysplasia ossificans profressiva","Genetics Home Reference", February 18, 2014.
- ↑ "Fibrodysoplasia ossificans profressiva", "Genetics Home Reference", February 18, 2014.
- ↑ 14.0 14.1 Hiroshi Kitoh et al. (2013). "Perhexiline maleate in the treatment of fibrodysplasia ossificans progressiva: an open-labeled clinical trial" 8. Orphanet Journal of Rare Diseases. p. 163.
- ↑ American Academy of Orthopaedic Surgeons (May 2006). "Fibrodysplasia Ossificans Progressiva (FOP)". orthoinfo.aaos.org. Retrieved 2011-10-07.
- ↑ Newton, M.C.; Allen, P.W.; Ryan, D.C. "Fibrodysplasia Ossificans Progressiva". British Journal of Anaesthesia. Retrieved October 25, 2011.
- ↑ BioInfoBank Library. "Squalamine: an aminosterol antibiotic from the shark". Lib.bioinfo.pl. Retrieved 2010-07-19.
- ↑ BBC News, "Shark therapy for bizarre bone disease," March 24, 1999
- ↑ "Squalamine Trial For The Treatment Of Fibrodysplasia Ossificans". Newsrx/Angiogenesis Weekly.
- ↑ "Article: Squalamine Trial For The Treatment Of Fibrodysplasia Ossificans Initiated... | AccessMyLibrary - Promoting library advocacy". AccessMyLibrary. 2002-02-08. Retrieved 2010-07-19.
- ↑ "Orphan drug designations and approvals list as of 09-03-2013" (PDF). USHRSA.
- ↑ "Oxfords Research Team". FOP Oxford Ball.
- ↑ Apirat Chaikuad et al. (2012-10-26). "Structure of the Bone Morphogenetic Protein Receptor ALK2 and Implications for Fibrodysplasia Ossificans Progressiva". The Journal of Biological Chemistry (J Biol Chem.) 287 (44): 36990–36998. doi:10.1074/jbc.M112.365932. PMC 3481300. PMID 22977237.
- ↑ Yu, P.B.; Deng, DY; Lai, CS; Hong, CC; Cuny, GD; Bouxsein, ML; Hong, DW; McManus, PM et al. (December 2008). "BMP type I receptor inhibition reduces heterotopic ossification". Nat Med. 14 (12): 1363–1369. doi:10.1038/nm.1888. PMC 2846458. PMID 19029982.
- ↑ J. W. Lowery and V. Rosen (2012). "Allele-Specific RNA Interference in FOP Silencing the FOP gene". Gene Therapy 19 (701–702): 701. doi:10.1038/gt.2011.190.
- ↑ "FOP Fact Sheet". ifopa. Retrieved 2013-12-12.
- ↑ Connor JM, Evans DA (1982). "Genetic aspects of fibrodysplasia ossificans progressiva". J. Med. Genet. 19 (1): 35–39. doi:10.1136/jmg.19.1.35. PMC 1048816. PMID 7069743.
- ↑ "What is FOP? » FOP Action - Fibrodysplasia Ossificans Progressiva Awareness". FOP Action. Retrieved 2013-05-24.
Further reading
- Cohen, MM; Howell, RE (October 1999). "Etiology of fibrous dysplasia and McCune-Albright syndrome". International journal of oral and maxillofacial surgery 28 (5): 366–71. PMID 10535539.
| ||||||||||||||||||||||||||||||||||||