FADD


- This article is about molecular biology. For other uses, see Fadd (disambiguation).
Fas-Associated protein with Death Domain (FADD), also called MORT1, is encoded by the FADD gene on the 11q13.3 region of chromosome 11 in humans.[1]
FADD is an adaptor protein that bridges members of the tumor necrosis factor receptor superfamily, such as the Fas-receptor, to procaspases 8 and 10 to form the death-inducing signaling complex (DISC) during apoptosis. As well as its most well known role in apoptosis, FADD has also been seen to play a role in other processes including proliferation, cell cycle regulation and development.
Structure
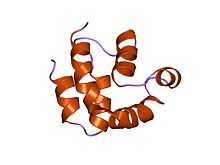
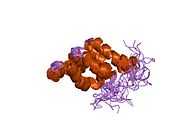
FADD is a 23 kDa protein, made up of 280 amino acids. It contains two main domains: a C terminal death domain (DD) and an N terminal death effector domain (DED). Each domain, although sharing very little sequence similarity, are structurally similar to one another, with each consisting of 6 α helices.[2][3] The DD of FADD binds to receptors such as the Fas receptor at the plasma membrane via their DD.[4] The interaction between the death domains are electrostatic interactions involving α helices 2 and 3 of the 6 helix domain.[5] The DED binds to the DED of intracellular molecules such as procaspase 8.[6] It is thought that this interaction occurs through hydrophobic interactions.[3]
Functions
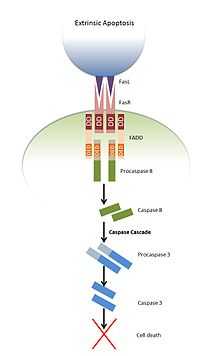
Extrinsic apoptosis
Upon stimulation by the Fas ligand, the Fas receptor trimerises. Many receptors, including Fas, contain a cytoplasmic DD and are therefore named death receptors. FADD binds to the DD of this trimeric structure via its death domain [4] resulting in unmasking of FADD's DED and subsequent recruitment of procaspase 8 and 10 via an interaction between the DEDs of both FADD and the procaspases.[7] This generates a complex known as the death inducing signalling complex (DISC).[8] Procaspase 8 and 10 are known as initiator caspases. These are inactive molecules, but when bought into close proximity with other procaspases of the same type, autocatalytic cleavage occurs at an aspartate residue within their own structures, resulting in an activated protein. This activated protein can then go on to cleave and activate further caspases, initiating the caspase cascade.[9] The activated caspases can go on to cleave intracellular proteins such as inhibitor of caspase-activated DNase (ICAD), which ultimately leads to apoptosis of the cell.[10]
Binding of TRAIL to death receptors four and five (DR4 and DR5) can lead to apoptosis by the same mechanism.[11]
Apoptosis can also be triggered by binding of a ligand to tumor necrosis factor receptor 1 (TNFR1); however, the mechanism by which this occurs is slightly more complex. Another DD-containing adaptor protein named TRADD, along with other proteins, binds to activated TNF1R, forming what is known as complex I. This results in activation of the NFκB pathway, which promotes cell survival. This complex is then internalised, and FADD binds to TRADD via an interaction of the DD’s of the two adapter proteins, forming what is known as complex II. FADD again recruits procaspase 8, which initiates the caspase cascade leading to apoptosis.[12]
Necroptosis
FADD also plays a role in regulating necroptosis, a process requiring the serine/threonine kinases, RIPK1 and RIPK3. Activated caspase 8 cleaves these kinases, inhibiting necroptosis. Since activation of caspase 8 requires FADD in order to bring the procaspase 8 molecules into close proximity to one another to facilitate their activation, FADD is required for negatively regulating necroptosis. In accordance, cells deficient in FADD induce necroptosis as they are unable to recruit and activate procaspase 8. FADD can also bind to RIPK1 and RIPK3 directly, however the significance of this interaction is currently unclear.[10]
Autophagic cell death
Autophagy is a process which allows cell survival under stressed conditions but can also lead to cell death.
Using its DD, FADD interacts with ATG5, a protein involved in autophagy. This interaction has been shown to be essential for autophagic cell death, which is induced by IFN-γ.[13]
In contrast, it has also been found to inhibit autophagic cell death and therefore promote cell survival. FADD binds to ATG5 in a complex which also contains ATG12, Caspase 8 and RIPK1. The formation of this complex is stimulated by autophagic signalling. Caspase 8 then cleaves RIPK1, leading to inhibition of this signalling, inhibiting cell death.[14]
Development
FADD knockout in mouse embryos is lethal, showing a role for FADD in embryonic development. This is thought to be due to abnormal development of the heart.[15] This abnormal heart development may be due to FADD dependent regulation of the NFκB pathway.[16]
FADD also plays a role in the development of the eyes of zebrafish.[17]
Cell cycle regulation
FADD is thought to have a role in regulating the cell cycle of T lymphocytes. This regulation is dependent on phosphorylation of FADD on Serine 194, which is carried out by Casein Kinase 1a (CKIα). This phosphorylated form of FADD is found mainly in the nucleus and the abundance of phosphorylated FADD increases significantly in the G2 phase of the cell cycle compared to the G1 phase where only very little can be detected. As it is found at the mitotic spindle during G2, it has been proposed to mediate the G2/M transition, however, the mechanism by which it does this it not yet known.[18]
Lymphocyte proliferation
FADD is essential for T cell proliferation when the T cell receptor is stimulated by antigen.[19] In contrast, FADD has no effect on the proliferation of B cells induced by stimulation of the B cell receptor. However, it is required for B cell proliferation induced by stimulation of TLR3 and TLR4.[20]
Inflammation
Activation of nuclear factor kappa B (NFκB) signalling leads to transcription of various proinflammatory cytokines as well as anti-apoptotic genes. It was found that NFκB signalling was inhibited in FADD-deficient cells after stimulation of the TNF-R1 or Fas receptors. This suggests a role of FADD in activation of the NFκB pathway. Conversely, FADD has also has a role in inhibition of this pathway. Normally, upon stimulation of the receptors TL4 or IL-1R1, the adaptor protein, MyD88, is recruited to the plasma membrane where is binds to IL-1 receptor associated Kinase (IRAK) via a DD-DD interaction. This activates a signalling pathway which results in translocation of NFκB to the nucleus, where it induces the transcription of the inflammatory cytokines. FADD can interfere with the interaction between MyD88 and IRAK, by binding to MyD88 via its DD and therefore this disrupts the cascade which would lead to NFκB translocation and inflammation.[21][22]
Other
FADD is required for an efficient antiviral response. Upon viral infection, FADD is needed to increase the levels of Irf7 a molecule which is needed for the production of IFN-α. IFN-α is a key molecule involved in the response against viruses.[23]
FADD is involved in the activation of the phosphatases which dephosphorylate and deactivate Protein Kinase C (PKC). Without FADD, PKC remains active and is able to continue signalling cascades leading to processes including cytoskeletal rearrangements and cell motility.[24]
Recent research has also shown that it may have a role in regulating glucose levels and the phosphorylated form of FADD is important for this function.[25]
Regulation
Subcellular localisation
FADD can be found in both the nucleus and cytoplasm of cells. Phosphorylation of Ser194 of FADD in humans (or Ser191 in mice) is thought to regulate its subcellular localisation. A nuclear localization sequence and nuclear export signal, both located in the DED of FADD, are also required for it to enter and exit the nucleus. Depending on its subcellular localisation, FADD can have different roles. In the cytoplasm, its main function is to induce apoptosis. However, in the nucleus, it can have the opposite effect and instead promote survival.[22][26]
c-FLIP
Cellular FLICE inhibitory protein (c-FLIP) is a regulatory protein which contains two DEDs. There are two isoforms of C-FLIP: C-FLIPS and FLIPL. It was originally thought to act as a negative regulator of apoptosis by binding to the DED of FADD and therefore preventing procaspase 8 from binding and inhibiting formation of the DISC.[27] However, it has been seen that both c-FLIP and procaspase 8 can be found at the same DISC.[28] Therefore it has been proposed that the presence of c-FLIP inhibits the close interaction of the procaspases to one another. Without this close proximity, the procaspases cannot be completely cleaved and remain in an inactive state.[27]
PKC
The activity of protein kinase C has a negative effect on Fas receptor mediated apoptosis. This is because it inhibits the recruitment of FADD to the receptor and so a DISC is not formed. It has been shown that by either increasing or decreasing the amount of PKC in T cells, more or less FADD is recruited to FasR respectively, when the FasR is stimulated.[29]
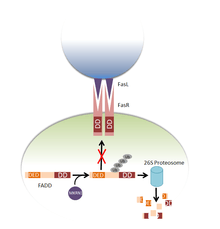
MKRN1
MKRN1 is an E3 ubiquitin ligase which negatively regulates FADD by targeting it for ubiquitin mediated degradation. In doing so, MKRN1 is able to control the level of apoptosis.[30]
Roles in inflammatory diseases
Increased levels of FADD were found in the leukocytes of patients with relapsing remitting multiple sclerosis, contributing to inflammation.[31] In rheumatoid arthritis, it is thought that stimulation of Fas receptors on macrophages, leads to formation of the FADD containing DISCs. Formation of these sequesters FADD away from MyD88 allowing MyD88 to interact with IRAK and induce the enhanced inflammation associated with this disease.[32]
Roles in Cancer
As FADD has such an important role in apoptosis, loss of FADD can give cancer cells a proliferative advantage as apoptosis would no longer be induced when the Fas receptors are stimulated.[22]
However, there is significant upregulation of FADD in head and neck squamous cell carcinoma. It is not yet clear what advantage this has on the cancer cells, but given FADDs roles in cell cycle regulation and cell survival, it likely that it may be related to this.[33] There are also elevated levels of FADD in non small cell lung cancer. FADD can be used as a prognosis marker for both of these diseases, with high levels of FADD being correlated with poor outcome.[34]
Therapeutic Target
Taxol is a drug used in anticancer therapies due to its ability to interfere with microtubule assembly, which leads to cell cycle arrest. FADD phosphorylated at Ser194 makes cells more sensitive to cell cycle arrest induced by taxol.[18] Taxol can also cause apoptosis of cells and this requires procaspase 10, which is activated by recruitment to FADD.[35]
It has been shown that the activation of JNK leads to the phosphorylation of FADD. Phosphorylated FADD can induce G2/M cell cycle arrest, potentially by increasing the stability of p53. Therefore drugs which can activate this pathway may have a therapeutic potential.[36] However, high levels of phosphorylated FADD have been correlated with a poor prognosis in many cancers such as that of the head and neck. This is likely to be due to its activation of the NF-κB pathway, which is antiapoptotic. Therefore, inhibition of FADD phosphorylation may be developed as a potential anti cancer strategy.[37]
Interactions
FADD has been seen to interact with Fas receptor,:[4]
See also
References
- ↑ Kim, P.K.M., Dutra, A.S., Chandrasekharappa, S.C., PUCK, J.M (1996). "Genomic structure and mapping of human FADD, an intracellular mediator of lymphocyte apoptosis". Journal of Immunology 157 (12): 5461–5466. PMID 8955195.
- ↑ Huang B, Eberstadt M, Olejniczak ET, Meadows RP, Fesik SW (1996). "NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain". Nature 384 (6610): 638–641. doi:10.1038/384638a0. PMID 8967952.
- ↑ 3.0 3.1 Eberstadt M, Huang B, Chen Z, Meadows RP, Ng SC, Zheng L et al. (1998). "NMR structure and mutagenesis of the FADD (Mort1) death-effector domain". Nature 392 (6679): 941–945. doi:10.1038/31972. PMID 9582077.
- ↑ 4.0 4.1 4.2 Boldin, M. P., Varfolomeev, E. E., Pancer, Z., Mett, I. L., Camonis, J. H. & Wallach, D. (1995). "A Novel Protein That Interacts with the Death Domain of Fas/APO1 Contains a Sequence Motif Related to the Death Domain". Journal of Biological Chemistry 270 (14): 7795–7798. doi:10.1074/jbc.270.14.7795. PMID 7536190.
- ↑ Jeong, E. J., Bang, S., Lee, T. H., Park, Y. I., Sim, W. S., Kim, K. S. (1999). "The solution structure of FADD death domain - Structural basis of death domain interactions of Fas and FADD". Journal of Biological Chemistry 274 (23): 16337–16342. doi:10.1074/jbc.274.23.16337. PMID 10347191.
- ↑ Boldin, M. P., Goncharov, T. M., Goltsev, Y. V., wallach, D. (1996). "Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death". Cell 85 (6): 803–815. doi:10.1016/s0092-8674(00)81265-9. PMID 8681376.
- ↑ 7.0 7.1 7.2 Kischkel FC, Lawrence DA, Tinel A, LeBlanc H, Virmani A, Schow P et al. (2001). "Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8". Journal of Biological Chemistry 276 (49): 46639–46646. doi:10.1074/jbc.M105102200. PMID 11583996.
- ↑ Pruul H, McDonald PJ (1995). "Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signalling complex (DISC) with the receptor". Embo Journal 14 (22): 5579–5588. PMC 394672.
- ↑ Weinlich R, Dillon CP, Green DR (2011). "Ripped to death". Trends in Cell Biology 21: 630–637. doi:10.1016/j.tcb.2011.09.002. PMC 3205316. PMID 21978761.
- ↑ 10.0 10.1 10.2 10.3 Lee, E.-W., Seo, J., Jeong, M., Lee, S., Song, J (2012). "The roles of FADD in extrinsic apoptosis and necroptosis". Bmb Reports 45 (9): 496–508. doi:10.5483/BMBRep.2012.45.9.186. PMID 23010170.
- ↑ 11.0 11.1 Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P et al. (2000). "TRAIL receptor-2 signals apoptosis through FADD and caspase-8". Nature Cell Biology 2 (4): 241–243. doi:10.1038/35008667. PMID 10783243.
- ↑ 12.0 12.1 Micheau, O., Tschopp, J. (2003). "Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes". Cell 114 (2): 181–190. doi:10.1016/s0092-8674(03)00521-x. PMID 12887920.
- ↑ 13.0 13.1 Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN et al. (2005). "Essential roles of Atg5 and FADD in autophagic cell death - Dissection of autophagic cell death into vacuole formation and cell death". Journal of Biological Chemistry 280 (21): 20722–20729. doi:10.1074/jbc.M413934200. PMID 15778222.
- ↑ Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA et al. (2008). ". FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells". Proceedings of the National Academy of Sciences of the United States of America 105: 16677–16682. doi:10.1073/pnas.0808597105. PMC 2575479. PMID 18946037.
- ↑ Yeh, W. C., De La Pompa, J. L., Mccurach, M. E., Shu, H. B., Elia, A. J., Shahinian, A., Ng, M., Wakeham, A., Khoo, W., Mitchell, K., El-Deiry, W. S., Lowe, S. W., Goeddel, D. V., Mak, T. W. (1998). "FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis". Science 279 (5358): 1954–1958. doi:10.1126/science.279.5358.1954. PMID 9506948.
- ↑ Sakamaki K, Takagi C, Kitayama A, Kurata T, Yamamoto TS, Chiba K et al. (2012). "Multiple functions of FADD in apoptosis, NF-kappa B-related signaling, and heart development in Xenopus embryos". Genes to Cells 17 (11): 875–896. doi:10.1111/gtc.12004. PMID 23025414.
- ↑ Gregory-Evans CY, Moosajee M, Hodges MD, Mackay DS, Game L, Vargesson N et al. (2007). "SNP genome scanning localizes oto-dental syndrome to chromosome 11q13 and microdeletions at this locus implicate FGF3 in dental and inner-ear disease and FADD in ocular coloboma". Human Molecular Genetics 16 (20): 2482–2493. doi:10.1093/hmg/ddm204. PMID 17656375.
- ↑ 18.0 18.1 18.2 Alappat EC, Feig C, Boyerinas B, Volkland J, Samuels M, Murmann AE et al. (2005). "Phosphorylation of FADD at serine 194 by CKI alpha regulates its nonapoptotic activities". Molecular Cell 19 (3): 321–332. doi:10.1016/j.molcel.2005.06.024. PMID 16061179.
- ↑ Zhang J, Cado D, Chen A, Kabra NH, Winoto A (1998). "Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1". Nature 392 (6673): 296–300. doi:10.1038/32681. PMID 9521326.
- ↑ Vander Elst P, van den Berg E, Pepermans H, vander Auwera L, Zeeuws R, Tourwe D et al. (2006). "The Fas-associated death domain protein is required in apoptosis and TLR-induced proliferative responses in B cells". Journal of Immunology 176: 6852–6861. doi:10.4049/jimmunol.176.11.6852. PMC 3110081.
- ↑ Wajant H, Haas E, Schwenzer R, Muhlenbeck F, Kreuz S, Schubert G et al. (2000). "Inhibition of death receptor-mediated gene induction by a cycloheximide-sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD)". Journal of Biological Chemistry 275 (32): 24357–24366. doi:10.1074/jbc.M000811200. PMID 10823821.
- ↑ 22.0 22.1 22.2 22.3 Tourneur L, Chiocchia G (2010). "FADD: a regulator of life and death". Trends in Immunology 31 (7): 260–269. doi:10.1016/j.it.2010.05.005. PMID 20576468.
- ↑ Balachandran, S., Venkataraman, T., Fisher, P. B., Barber, G. N (2007). "Fas-associated death domain-containing protein-mediated antiviral innate immune signaling involves the regulation of Irf7". Journal of Immunology 178 (4): 2429–2439. doi:10.4049/jimmunol.178.4.2429. PMID 17277150.
- ↑ Cheng W, Wang L, Zhang R, Du P, Yang B, Zhuang H et al. (2012). "Regulation of Protein Kinase C Inactivation by Fas-associated Protein with Death Domain". Journal of Biological Chemistry 287: 26126–26135. doi:10.1074/jbc.M112.342170. PMC 3406696. PMID 22582393.
- ↑ Yao C, Zhuang H, Du P, Cheng W, Yang B, Guan S et al. (2013). "Domain-containing Protein (FADD) Phosphorylation in Regulating Glucose Homeostasis: from Proteomic Discovery to Physiological Validation". Molecular & Cellular Proteomics 12 (10): 2689–2700. doi:10.1074/mcp.M113.029306. PMID 23828893.
- ↑ Gómez-Angelats M, Cidlowski JA (2003). "Molecular evidence for the nuclear localization of FADD". Cell Death and Differentiation 10 (7): 791–797. doi:10.1038/sj.cdd.4401237. PMID 12815462.
- ↑ 27.0 27.1 Krueger A, Baumann S, Krammer PH, Kirchhoff S (2001). "FLICE-inhibitory proteins: Regulators of death receptor-mediated apoptosis". Molecular and Cellular Biology 21: 8247–8254. doi:10.1128/mcb.21.24.8247-8254.2001. PMC 99990. PMID 11713262.
- ↑ 28.0 28.1 Scaffidi, C., Schmitz, I., Krammer, P. H., Peter, M. E. (1999). "The role of c-FLIP in modulation of CD95-induced apoptosis". Journal of Biological Chemistry 274 (3): 1541–1548. doi:10.1074/jbc.274.3.1541. PMID 9880531.
- ↑ Gómez-Angelats M, Cidlowski JA (2001). "Protein kinase C regulates FADD recruitment and death-inducing signaling complex formation in Fas/CD95-induced apoptosis". Journal of Biological Chemistry 276 (48): 44944–44952. doi:10.1074/jbc.M104919200. PMID 11581255.
- ↑ 30.0 30.1 Lee EW, Kim JH, Ahn YH, Seo J, Ko A, Jeong M et al. (2012). "Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis". Nature Communications 3: 978. doi:10.1038/ncomms1981. PMID 22864571.
- ↑ Reuss R, Mistarz M, Mirau A, Kraus J, Bödeker RH, Oschmann P (2014). "FADD is upregulated in relapsing remitting multiple". Neuroimmunomodulation 1 (5): 221–225. doi:10.1159/000356522. PMID 24603611.
- ↑ Ma Y, Liu H, Tu-Rapp H, Thiesen HJ, Ibrahim SM, Cole SM et al. (2004). "Fas ligation on macrophages enhances IL-1R1-Toll-like receptor 4 signaling and promotes chronic inflammation". Nature Immunology 5 (4): 380–387. doi:10.1038/ni1054. PMID 15004557.
- ↑ Pattje WJ, Melchers LJ, Slagter-Menkema L, Mastik MF, Schrijvers ML, Gibcus JH et al. (2013). "FADD expression is associated with regional and distant metastasis in squamous cell carcinoma of the head and neck". Histopathology 63 (2): 263–270. doi:10.1111/his.12174. PMID 23763459.
- ↑ Cimino Y, Costes A, Damotte D, Validire P, Mistou S, Cagnard N et al. (2012). "FADD protein release mirrors the development and aggressiveness of human non-small cell lung cancer". British Journal of Cancer 106 (12): 1989–1996. doi:10.1038/bjc.2012.196. PMC 3388563. PMID 22669160.
- ↑ Park SJ, Wu CH, Gordon JD, Zhong X, Emami A, Safa AR (2004). "Taxol induces caspase-10-dependent apoptosis". Journal of Biological Chemistry 279 (49): 51057–51067. doi:10.1074/jbc.M406543200. PMID 15452117.
- ↑ Matsuyoshi S, Shimada K, Nakamura M, Ishida E, Konishi N (2006). "FADD phosphorylation is critical for cell cycle regulation in breast cancer cells". British Journal of Cancer 94 (4): 532–539. doi:10.1038/sj.bjc.6602955. PMC 2361184. PMID 16450001.
- ↑ Schinske KA, Nyati S, Khan AP, Williams TM, Johnson TD, Ross BD et al. (2011). "A Novel Kinase Inhibitor of FADD Phosphorylation Chemosensitizes through the Inhibition of NF-kappa B". Molecular Cancer Therapeutics 10: 1807–1817. doi:10.1158/1535-7163.mct-11-0362. PMC 3191281. PMID 21859840.
- ↑ Buechler C, Bared SM, Aslanidis C, Ritter M, Drobnik W, Schmitz G (Nov 2002). "Molecular and functional interaction of the ATP-binding cassette transporter A1 with Fas-associated death domain protein". J. Biol. Chem. 277 (44): 41307–10. doi:10.1074/jbc.C200436200. PMID 12235128.
- ↑ Roth W, Stenner-Liewen F, Pawlowski K, Godzik A, Reed JC (Mar 2002). "Identification and characterization of DEDD2, a death effector domain-containing protein". J. Biol. Chem. 277 (9): 7501–8. doi:10.1074/jbc.M110749200. PMID 11741985.
- ↑ Screaton RA, Kiessling S, Sansom OJ, Millar CB, Maddison K, Bird A et al. (Apr 2003). "Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: a potential link between genome surveillance and apoptosis". Proc. Natl. Acad. Sci. U.S.A. 100 (9): 5211–6. doi:10.1073/pnas.0431215100. PMC 154324. PMID 12702765.
- ↑ Stilo R, Liguoro D, di Jeso B, Leonardi A, Vito P (Apr 2003). "The alpha-chain of the nascent polypeptide-associated complex binds to and regulates FADD function". Biochem. Biophys. Res. Commun. 303 (4): 1034–41. doi:10.1016/s0006-291x(03)00487-x. PMID 12684039.
- ↑ Condorelli G, Vigliotta G, Cafieri A, Trencia A, Andalò P, Oriente F et al. (Aug 1999). "PED/PEA-15: an anti-apoptotic molecule that regulates FAS/TNFR1-induced apoptosis". Oncogene 18 (31): 4409–15. doi:10.1038/sj.onc.1202831. PMID 10442631.
Further reading
- Bolze A, Byun M, McDonald D, Morgan NV, Abhyankar A, Premkumar L et al. (2010). "Whole-Exome-Sequencing-Based Discovery of Human FADD Deficiency". Journal of Human Genetics 87: 873–881. doi:10.1016/j.ajhg.2010.10.028. PMC 2997374. PMID 21109225.
- Werner MH, Wu C, Walsh CM (2006). "Emerging roles for the death adaptor FADD in death receptor avidity and cell cycle regulation". Cell Cycle 5 (20): 2332–2338. doi:10.4161/cc.5.20.3385. PMID 17102623.
- Yu JW, Shi Y (2008). "FLIP and the death effector domain family". Oncogene 27 (48): 6216–6227. doi:10.1038/onc.2008.299. PMID 18931689.
- Bhojani MS, Chen G, Ross BD, Beer DG, Rehemtulla A (2007). "Nuclear localized phosphorylated FADD induces cell proliferation and is associated with aggressive lung cancer". Cell Cycle 4 (11): 1478–81. doi:10.4161/cc.4.11.2188. PMID 16258269.
External links
- Fas-Associating Protein with Death Domain at the US National Library of Medicine Medical Subject Headings (MeSH)
| |||||||||||||||||



| ||||||||||||||||||||||||||||||||||||||||||||||