Epigenetics
In genetics, epigenetics is the study of cellular and physiological trait variations that are not caused by changes in the DNA sequence; epigenetics describes the study of dynamic alterations in the transcriptional potential of a cell. These alterations may or may not be heritable, although the use of the term epigenetic to describe processes that are not heritable is controversial.[1] Unlike genetics based on changes to the DNA sequence (the genotype), the changes in gene expression or cellular phenotype of epigenetics have other causes, thus use of the term epi- (Greek: επί- over, outside of, around) -genetics.[2][3]
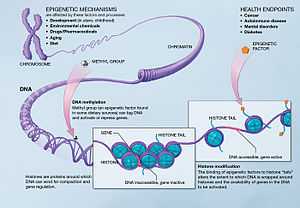
The term also refers to the changes themselves: functionally relevant changes to the genome that do not involve a change in the nucleotide sequence. Examples of mechanisms that produce such changes are DNA methylation and histone modification, each of which alters how genes are expressed without altering the underlying DNA sequence. Gene expression can be controlled through the action of repressor proteins that attach to silencer regions of the DNA. These epigenetic changes may last through cell divisions for the duration of the cell's life, and may also last for multiple generations even though they do not involve changes in the underlying DNA sequence of the organism;[4] instead, non-genetic factors cause the organism's genes to behave (or "express themselves") differently.[5]
One example of an epigenetic change in eukaryotic biology is the process of cellular differentiation. During morphogenesis, totipotent stem cells become the various pluripotent cell lines of the embryo, which in turn become fully differentiated cells. In other words, as a single fertilized egg cell – the zygote – continues to divide, the resulting daughter cells change into all the different cell types in an organism, including neurons, muscle cells, epithelium, endothelium of blood vessels, etc., by activating some genes while inhibiting the expression of others.[6]
Definitions of term
Historical usage
Epigenetics (as in "epigenetic landscape") was coined by C. H. Waddington in 1942 as a portmanteau of the words epigenesis and genetics.[7] Epigenesis is an old[8] word that has more recently been used (see preformationism for historical background) to describe the differentiation of cells from their initial totipotent state in embryonic development. When Waddington coined the term the physical nature of genes and their role in heredity was not known; he used it as a conceptual model of how genes might interact with their surroundings to produce a phenotype; he used the phrase "epigenetic landscape" as a metaphor for biological development. Waddington held that cell fates were established in development much like a marble rolls down to the point of lowest local elevation.[9] Waddington suggested visualising increasing irreversibility of cell type differentiation as ridges rising between the valleys where the marbles (cells) are travelling.[10] In recent times Waddington's notion of the epigenetic landscape has been rigorously formalized in the context of the systems dynamics state approach to the study of cell-fate.[11][12] Cell-fate determination is predicted to exhibit certain dynamics, such as attractor-convergence (the attractor can be an equilibrium point, limit cycle or strange attractor) or oscillatory.[12]
The term "epigenetics" has also been used in developmental psychology to describe psychological development as the result of an ongoing, bi-directional interchange between heredity and the environment.[13] Interactivist ideas of development have been discussed in various forms and under various names throughout the 19th and 20th centuries. An early version was proposed, among the founding statements in embryology, by Karl Ernst von Baer and popularized by Ernst Haeckel. A radical epigenetic view (physiological epigenesis) was developed by Paul Wintrebert. Another variation, probabilistic epigenesis, was presented by Gilbert Gottlieb in 2003.[14] This view encompasses all of the possible developing factors on an organism and how they not only influence the organism and each other, but how the organism also influences its own development.
The developmental psychologist Erik Erikson used the term epigenetic principle in his book Identity: Youth and Crisis (1968), and used it to encompass the notion that we develop through an unfolding of our personality in predetermined stages, and that our environment and surrounding culture influence how we progress through these stages. This biological unfolding in relation to our socio-cultural settings is done in stages of psychosocial development, where "progress through each stage is in part determined by our success, or lack of success, in all the previous stages."[15][16][17]
Contemporary usage
Robin Holliday defined epigenetics as "the study of the mechanisms of temporal and spatial control of gene activity during the development of complex organisms."[18] Thus epigenetic can be used to describe anything other than DNA sequence that influences the development of an organism.
The more recent usage of the word in science has a stricter definition. It is, as defined by Arthur Riggs and colleagues, "the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence."[19] The Greek prefix epi- in epigenetics implies features that are "on top of" or "in addition to" genetics; thus epigenetic traits exist on top of or in addition to the traditional molecular basis for inheritance.
The term "epigenetics", however, has been used to describe processes which have not been demonstrated to be heritable such as histone modification; there are therefore attempts to redefine it in broader terms that would avoid the constraints of requiring heritability. For example, Sir Adrian Bird defined epigenetics as "the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states."[4] This definition would be inclusive of transient modifications associated with DNA repair or cell-cycle phases as well as stable changes maintained across multiple cell generations, but exclude others such as templating of membrane architecture and prions unless they impinge on chromosome function. Such redefinitions however are not universally accepted and are still subject to dispute.[1] The NIH "Roadmap Epigenomics Project," ongoing as of 2013, uses the following definition: "...For purposes of this program, epigenetics refers to both heritable changes in gene activity and expression (in the progeny of cells or of individuals) and also stable, long-term alterations in the transcriptional potential of a cell that are not necessarily heritable."[20]
In 2008, a consensus definition of the epigenetic trait, "stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence", was made at a Cold Spring Harbor meeting.[21]
The similarity of the word to "genetics" has generated many parallel usages. The "epigenome" is a parallel to the word "genome", referring to the overall epigenetic state of a cell, and epigenomics refers to more global analyses of epigenetic changes across the entire genome.[20] The phrase "genetic code" has also been adapted—the "epigenetic code" has been used to describe the set of epigenetic features that create different phenotypes in different cells. Taken to its extreme, the "epigenetic code" could represent the total state of the cell, with the position of each molecule accounted for in an epigenomic map, a diagrammatic representation of the gene expression, DNA methylation and histone modification status of a particular genomic region. More typically, the term is used in reference to systematic efforts to measure specific, relevant forms of epigenetic information such as the histone code or DNA methylation patterns.
Molecular basis
Epigenetic changes can modify the activation of certain genes, but not the sequence of DNA. Additionally, the chromatin proteins associated with DNA may be activated or silenced. This is why the differentiated cells in a multicellular organism express only the genes that are necessary for their own activity. Epigenetic changes are preserved when cells divide. Most epigenetic changes only occur within the course of one individual organism's lifetime, but, if gene inactivation occurs in a sperm or egg cell that results in fertilization, then some epigenetic changes can be transferred to the next generation.[22] This raises the question of whether or not epigenetic changes in an organism can alter the basic structure of its DNA (see Evolution, below), a form of Lamarckism.
Specific epigenetic processes include paramutation, bookmarking, imprinting, gene silencing, X chromosome inactivation, position effect, reprogramming, transvection, maternal effects, the progress of carcinogenesis, many effects of teratogens, regulation of histone modifications and heterochromatin, and technical limitations affecting parthenogenesis and cloning.
DNA damage can also cause epigenetic changes.[23][24][25] DNA damages are very frequent, occurring on average about 10,000 times a day per cell of the human body (see DNA damage (naturally occurring)). These damages are largely repaired, but at the site of a DNA repair, epigenetic changes can remain.[26] In particular, a double strand break in DNA can initiate unprogrammed epigenetic gene silencing both by causing DNA methylation as well as by promoting silencing types of histone modifications (chromatin remodeling) (see next section).[27] In addition, the enzyme Parp1 (poly(ADP)-ribose polymerase) and its product poly(ADP)-ribose (PAR) accumulate at sites of DNA damage as part of a repair process.[28] This accumulation, in turn, directs recruitment and activation of the chromatin remodeling protein ALC1 that can cause nucleosome remodeling.[29] Nucleosome remodeling has been found to cause, for instance, epigenetic silencing of DNA repair gene MLH1.[19][30] DNA damaging chemicals, such as benzene, hydroquinone, styrene, carbon tetrachloride and trichloroethylene, cause considerable hypomethylation of DNA, some through the activation of oxidative stress pathways.[31]
Foods are known to alter the epigenetics of rats on different diets.[32] Some food components epigenetically increase the levels of DNA repair enzymes such as MGMT and MLH1[33] and p53.[34][35] Other food components can reduce DNA damage, such as soy isoflavones[36][37] and bilberry anthocyanins.[38]
Epigenetic research uses a wide range of molecular biologic techniques to further our understanding of epigenetic phenomena, including chromatin immunoprecipitation (together with its large-scale variants ChIP-on-chip and ChIP-Seq), fluorescent in situ hybridization, methylation-sensitive restriction enzymes, DNA adenine methyltransferase identification (DamID) and bisulfite sequencing. Furthermore, the use of bioinformatic methods is playing an increasing role (computational epigenetics).
Computer simulations and molecular dynamics approaches revealed the atomistic motions associated with the molecular recognition of the histone tail through an allosteric mechanism.[39]
Mechanisms
Several types of epigenetic inheritance systems may play a role in what has become known as cell memory,[40] note however that not all of these are universally accepted to be examples of epigenetics.
DNA methylation and chromatin remodeling
Because DNA methylation and chromatin remodeling play such a central role in many types of epigenic inheritance, the word "epigenetics" is sometimes used as a synonym for these processes. However, this can be misleading. Chromatin remodeling is not always inherited, and not all epigenetic inheritance involves chromatin remodeling.[41]

Because the phenotype of a cell or individual is affected by which of its genes are transcribed, heritable transcription states can give rise to epigenetic effects. There are several layers of regulation of gene expression. One way that genes are regulated is through the remodeling of chromatin. Chromatin is the complex of DNA and the histone proteins with which it associates. If the way that DNA is wrapped around the histones changes, gene expression can change as well. Chromatin remodeling is accomplished through two main mechanisms:
- The first way is post translational modification of the amino acids that make up histone proteins. Histone proteins are made up of long chains of amino acids. If the amino acids that are in the chain are changed, the shape of the histone might be modified. DNA is not completely unwound during replication. It is possible, then, that the modified histones may be carried into each new copy of the DNA. Once there, these histones may act as templates, initiating the surrounding new histones to be shaped in the new manner. By altering the shape of the histones around them, these modified histones would ensure that a lineage-specific transcription program is maintained after cell division.
- The second way is the addition of methyl groups to the DNA, mostly at CpG sites, to convert cytosine to 5-methylcytosine. 5-Methylcytosine performs much like a regular cytosine, pairing with a guanine in double-stranded DNA. However, some areas of the genome are methylated more heavily than others, and highly methylated areas tend to be less transcriptionally active, through a mechanism not fully understood. Methylation of cytosines can also persist from the germ line of one of the parents into the zygote, marking the chromosome as being inherited from one parent or the other (genetic imprinting).
Mechanisms of heritability of histone state are not well understood; however, much is known about the mechanism of heritability of DNA methylation state during cell division and differentiation. Heritability of methylation state depends on certain enzymes (such as DNMT1) that have a higher affinity for 5-methylcytosine than for cytosine. If this enzyme reaches a "hemimethylated" portion of DNA (where 5-methylcytosine is in only one of the two DNA strands) the enzyme will methylate the other half.
Although histone modifications occur throughout the entire sequence, the unstructured N-termini of histones (called histone tails) are particularly highly modified. These modifications include acetylation, methylation, ubiquitylation, phosphorylation, sumoylation, ribosylation and citrullination. Acetylation is the most highly studied of these modifications. For example, acetylation of the K14 and K9 lysines of the tail of histone H3 by histone acetyltransferase enzymes (HATs) is generally related to transcriptional competence.
One mode of thinking is that this tendency of acetylation to be associated with "active" transcription is biophysical in nature. Because it normally has a positively charged nitrogen at its end, lysine can bind the negatively charged phosphates of the DNA backbone. The acetylation event converts the positively charged amine group on the side chain into a neutral amide linkage. This removes the positive charge, thus loosening the DNA from the histone. When this occurs, complexes like SWI/SNF and other transcriptional factors can bind to the DNA and allow transcription to occur. This is the "cis" model of epigenetic function. In other words, changes to the histone tails have a direct effect on the DNA itself.
Another model of epigenetic function is the "trans" model. In this model, changes to the histone tails act indirectly on the DNA. For example, lysine acetylation may create a binding site for chromatin-modifying enzymes (or transcription machinery as well). This chromatin remodeler can then cause changes to the state of the chromatin. Indeed, a bromodomain — a protein domain that specifically binds acetyl-lysine — is found in many enzymes that help activate transcription, including the SWI/SNF complex. It may be that acetylation acts in this and the previous way to aid in transcriptional activation.
The idea that modifications act as docking modules for related factors is borne out by histone methylation as well. Methylation of lysine 9 of histone H3 has long been associated with constitutively transcriptionally silent chromatin (constitutive heterochromatin). It has been determined that a chromodomain (a domain that specifically binds methyl-lysine) in the transcriptionally repressive protein HP1 recruits HP1 to K9 methylated regions. One example that seems to refute this biophysical model for methylation is that tri-methylation of histone H3 at lysine 4 is strongly associated with (and required for full) transcriptional activation. Tri-methylation in this case would introduce a fixed positive charge on the tail.
It has been shown that the histone lysine methyltransferase (KMT) is responsible for this methylation activity in the pattern of histones H3 & H4. This enzyme utilizes a catalytically active site called the SET domain (Suppressor of variegation, Enhancer of zeste, Trithorax). The SET domain is a 130-amino acid sequence involved in modulating gene activities. This domain has been demonstrated to bind to the histone tail and causes the methylation of the histone.[42]
Differing histone modifications are likely to function in differing ways; acetylation at one position is likely to function differently from acetylation at another position. Also, multiple modifications may occur at the same time, and these modifications may work together to change the behavior of the nucleosome. The idea that multiple dynamic modifications regulate gene transcription in a systematic and reproducible way is called the histone code, although the idea that histone state can be read linearly as a digital information carrier has been largely debunked. One of the best-understood systems that orchestrates chromatin-based silencing is the SIR protein based silencing of the yeast hidden mating type loci HML and HMR.
DNA methylation frequently occurs in repeated sequences, and helps to suppress the expression and mobility of 'transposable elements':[43] Because 5-methylcytosine can be spontaneously deaminated (replacing nitrogen by oxygen) to thymidine, CpG sites are frequently mutated and become rare in the genome, except at CpG islands where they remain unmethylated. Epigenetic changes of this type thus have the potential to direct increased frequencies of permanent genetic mutation. DNA methylation patterns are known to be established and modified in response to environmental factors by a complex interplay of at least three independent DNA methyltransferases, DNMT1, DNMT3A, and DNMT3B, the loss of any of which is lethal in mice.[44] DNMT1 is the most abundant methyltransferase in somatic cells,[45] localizes to replication foci,[46] has a 10–40-fold preference for hemimethylated DNA and interacts with the proliferating cell nuclear antigen (PCNA).[47]
By preferentially modifying hemimethylated DNA, DNMT1 transfers patterns of methylation to a newly synthesized strand after DNA replication, and therefore is often referred to as the ‘maintenance' methyltransferase.[48] DNMT1 is essential for proper embryonic development, imprinting and X-inactivation.[44][49] To emphasize the difference of this molecular mechanism of inheritance from the canonical Watson-Crick base-pairing mechanism of transmission of genetic information, the term 'Epigenetic templating' was introduced.[50] Furthermore, in addition to the maintenance and transmission of methylated DNA states, the same principle could work in the maintenance and transmission of histone modifications and even cytoplasmic (structural) heritable states.[51]
Histones H3 and H4 can also be manipulated through demethylation using histone lysine demethylase (KDM). This recently identified enzyme has a catalytically active site called the Jumonji domain (JmjC). The demethylation occurs when JmjC utilizes multiple cofactors to hydroxylate the methyl group, thereby removing it. JmjC is capable of demethylating mono-, di-, and tri-methylated substrates.[52]
Chromosomal regions can adopt stable and heritable alternative states resulting in bistable gene expression without changes to the DNA sequence. Epigenetic control is often associated with alternative covalent modifications of histones.[53] The stability and heritability of states of larger chromosomal regions are suggested to involve positive feedback where modified nucleosomes recruit enzymes that similarly modify nearby nucleosomes.[54] A simplified stochastic model for this type of epigenetics is found here.[55][56]
It has been suggested that chromatin-based transcriptional regulation could be mediated by the effect of small RNAs. Small interfering RNAs can modulate transcriptional gene expression via epigenetic modulation of targeted promoters.[57]
RNA transcripts and their encoded proteins
Sometimes a gene, after being turned on, transcribes a product that (directly or indirectly) maintains the activity of that gene. For example, Hnf4 and MyoD enhance the transcription of many liver- and muscle-specific genes, respectively, including their own, through the transcription factor activity of the proteins they encode. RNA signalling includes differential recruitment of a hierarchy of generic chromatin modifying complexes and DNA methyltransferases to specific loci by RNAs during differentiation and development.[58] Other epigenetic changes are mediated by the production of different splice forms of RNA, or by formation of double-stranded RNA (RNAi). Descendants of the cell in which the gene was turned on will inherit this activity, even if the original stimulus for gene-activation is no longer present. These genes are often turned on or off by signal transduction, although in some systems where syncytia or gap junctions are important, RNA may spread directly to other cells or nuclei by diffusion. A large amount of RNA and protein is contributed to the zygote by the mother during oogenesis or via nurse cells, resulting in maternal effect phenotypes. A smaller quantity of sperm RNA is transmitted from the father, but there is recent evidence that this epigenetic information can lead to visible changes in several generations of offspring.[59]
MicroRNAs
MicroRNAs (miRNAs) are members of non-coding RNAs that range in size from 17 to 25 nucleotides. miRNAs regulate a large variety of biological functions in plants and animals.[60] So far, in 2013, about 2000 miRNAs have been discovered in humans and these can be found online in an miRNA database.[61] Each miRNA expressed in a cell may target about 100 to 200 messenger RNAs that it downregulates.[62] Most of the downregulation of mRNAs occurs by causing the decay of the targeted mRNA, while some downregulation occurs at the level of translation into protein.[63]
It appears that about 60% of human protein coding genes are regulated by miRNAs.[64] Many miRNAs are epigenetically regulated. About 50% of miRNA genes are associated with CpG islands,[60] that may be repressed by epigenetic methylation. Transcription from methylated CpG islands is strongly and heritably repressed.[65] Other miRNAs are epigenetically regulated by either histone modifications or by combined DNA methylation and histone modification.[60]
mRNA
In 2011, it was demonstrated that the methylation of mRNA plays a critical role in human energy homeostasis. The obesity-associated FTO gene is shown to be able to demethylate N6-methyladenosine in RNA.[66][67]
sRNAs
sRNAs are small (50–250 nucleotides), highly structured, non-coding RNA fragments found in bacteria. They control gene expression including virulence genes in pathogens and are viewed as new targets in the fight against drug-resistant bacteria.[68] They play an important role in many biological processes, binding to mRNA and protein targets in prokaryotes. Their phylogenetic analyses, for example through sRNA–mRNA target interactions or protein binding properties, are used to build comprehensive databases.[69] sRNA-gene maps based on their targets in microbial genomes are also constructed.[70]
Prions
Prions are infectious forms of proteins. In general, proteins fold into discrete units that perform distinct cellular functions, but some proteins are also capable of forming an infectious conformational state known as a prion. Although often viewed in the context of infectious disease, prions are more loosely defined by their ability to catalytically convert other native state versions of the same protein to an infectious conformational state. It is in this latter sense that they can be viewed as epigenetic agents capable of inducing a phenotypic change without a modification of the genome.[71]
Fungal prions are considered by some to be epigenetic because the infectious phenotype caused by the prion can be inherited without modification of the genome. PSI+ and URE3, discovered in yeast in 1965 and 1971, are the two best studied of this type of prion.[72][73] Prions can have a phenotypic effect through the sequestration of protein in aggregates, thereby reducing that protein's activity. In PSI+ cells, the loss of the Sup35 protein (which is involved in termination of translation) causes ribosomes to have a higher rate of read-through of stop codons, an effect that results in suppression of nonsense mutations in other genes.[74] The ability of Sup35 to form prions may be a conserved trait. It could confer an adaptive advantage by giving cells the ability to switch into a PSI+ state and express dormant genetic features normally terminated by stop codon mutations.[75][76][77][78]
Structural inheritance systems
In ciliates such as Tetrahymena and Paramecium, genetically identical cells show heritable differences in the patterns of ciliary rows on their cell surface. Experimentally altered patterns can be transmitted to daughter cells. It seems existing structures act as templates for new structures. The mechanisms of such inheritance are unclear, but reasons exist to assume that multicellular organisms also use existing cell structures to assemble new ones.[79][80][81]
Functions and consequences
Development
Somatic epigenetic inheritance through epigenetic modifications, particularly through DNA methylation and chromatin remodeling, is very important in the development of multicellular eukaryotic organisms. The genome sequence is static (with some notable exceptions), but cells differentiate into many different types, which perform different functions, and respond differently to the environment and intercellular signalling. Thus, as individuals develop, morphogens activate or silence genes in an epigenetically heritable fashion, giving cells a memory. In mammals, most cells terminally differentiate, with only stem cells retaining the ability to differentiate into several cell types ("totipotency" and "multipotency"). In mammals, some stem cells continue producing new differentiated cells throughout life, such as in neurogenesis, but mammals are not able to respond to loss of some tissues, for example, the inability to regenerate limbs, which some other animals are capable of. Epigenetic modifications regulate the transition from neural neural stem cells to glial progenitor cells (for example, differentiation into oligodendrocytes is regulated by the deacetylation and methylation of histones.[82] Unlike animals, plant cells do not terminally differentiate, remaining totipotent with the ability to give rise to a new individual plant. While plants do utilise many of the same epigenetic mechanisms as animals, such as chromatin remodeling, it has been hypothesised that some kinds of plant cells do not use or require "cellular memories", resetting their gene expression patterns using positional information from the environment and surrounding cells to determine their fate.[83]
Epigenetics can be divided into predetermined and probabilistic epigenesis. Predetermined epigenesis is a unidirectional movement from structural development in DNA to the functional maturation of the protein. "Predetermined" here means that development is scripted and predictable. Probabilistic epigenesis on the other hand is a bidirectional structure-function development with experiences and external molding development.[84]
Medicine
Epigenetics has many and varied potential medical applications as it tends to be multidimensional in nature.[85]
Congenital genetic disease is well understood and it is clear that epigenetics can play a role, for example, in the case of Angelman syndrome and Prader-Willi syndrome. These are normal genetic diseases caused by gene deletions or inactivation of the genes, but are unusually common because individuals are essentially hemizygous because of genomic imprinting, and therefore a single gene knock out is sufficient to cause the disease, where most cases would require both copies to be knocked out.[86]
Evolution
Epigenetics can impact evolution when epigenetic changes are heritable. A sequestered germ line or Weismann barrier is specific to animals, and epigenetic inheritance is more common in plants and microbes. Eva Jablonka and Marion Lamb have argued that these effects may require enhancements to the standard conceptual framework of the modern evolutionary synthesis.[87][88] Other evolutionary biologists have incorporated epigenetic inheritance into population genetics models[89] or are openly skeptical.[90]
Two important ways in which epigenetic inheritance can be different from traditional genetic inheritance, with important consequences for evolution, are that rates of epimutation can be much faster than rates of mutation[91] and the epimutations are more easily reversible.[92] An epigenetically inherited element such as the PSI+ system can act as a "stop-gap", good enough for short-term adaptation that allows the lineage to survive for long enough for mutation and/or recombination to genetically assimilate the adaptive phenotypic change.[93] The existence of this possibility increases the evolvability of a species.
Current research findings and examples of effects
Epigenetic changes have been observed to occur in response to environmental exposure—for example, mice given some dietary supplements have epigenetic changes affecting expression of the agouti gene, which affects their fur color, weight, and propensity to develop cancer.[94][95]
One study indicates that traumatic experiences can produce fearful memories which are passed to future generations via epigenetics. A study carried out on mice in 2013 found that mice could produce offspring which had an aversion to certain items which had been the source of negative experiences for their ancestors.[96][97] Reports stated that:
For the study, author Brian Dias and co-author Kerry Ressler trained mice, using foot shocks, to fear an odour that resembles cherry blossoms. Later, they tested the extent to which the animals' offspring startled when exposed to the same smell. The younger generation had not even been conceived when their fathers underwent the training, and had never smelt the odour before the experiment.The offspring of trained mice were "able to detect and respond to far less amounts of odour... suggesting they are more sensitive" to it, Ressler told AFP of the findings published in the journal Nature Neuroscience. They did not react the same way to other odours, and compared to the offspring of non-trained mice, their reaction to the cherry blossom whiff was about 200 percent stronger, he said.
The scientists then looked at a gene, M71, that governs the functioning of an odour receptor in the nose that responds specifically to the cherry blossom smell. The gene, inherited through the sperm of trained mice, had undergone no change to its DNA encoding, the team found. But the gene did carry epigenetic marks that could alter its behaviour and cause it to be "expressed more" in descendants, said Dias. This in turn caused a physical change in the brains of the trained mice, their sons and grandsons, who all had a larger glomerulus—a section in the olfactory (smell) unit of the brain.
This study received several criticisms. In a paper in GENETICS, author Gregory Francis cited the study's low statistical power as evidence of some irregularity such as bias in reporting results.[98] Due to limits of sample size, there is a probability that an effect will not be demonstrated to within statistical significance even if it exists. Francis calculated the probability that all the experiments reported would show positive results if an identical protocol was followed, assuming the claimed effects exist, to be 0.4%. Another criticism was that the authors did not indicate which mice were siblings, and treated all of the mice as statistically independent.[99] Dias and Ressler responded by pointing out negative results in the paper's appendix that Francis did not use in his calculations, and by saying they would track which mice were siblings in the future.[100]
In the case of humans with different environmental exposures, monozygotic (identical) twins were epigenetically indistinguishable during their early years, while older twins had remarkable differences in the overall content and genomic distribution of 5-methylcytosine DNA and histone acetylation. The twin pairs who had spent less of their lifetime together and/or had greater differences in their medical histories were those who showed the largest differences in their levels of 5-methylcytosine DNA and acetylation of histones H3 and H4.[101]
More than 100 cases of transgenerational epigenetic inheritance phenomena have been reported in a wide range of organisms, including prokaryotes, plants, and animals.[102] For instance, Mourning Cloak butterflies will change color through hormone changes in response to experimentation of varying temperatures.[103]
Recent analyses have suggested that members of the APOBEC/AID family of cytosine deaminases are capable of simultaneously mediating genetic and epigenetic inheritance using similar molecular mechanisms.[104]
Epigenetic effects in humans
Genomic imprinting and related disorders
Some human disorders are associated with genomic imprinting, a phenomenon in mammals where the father and mother contribute different epigenetic patterns for specific genomic loci in their germ cells.[105] The best-known case of imprinting in human disorders is that of Angelman syndrome and Prader-Willi syndrome—both can be produced by the same genetic mutation, chromosome 15q partial deletion, and the particular syndrome that will develop depends on whether the mutation is inherited from the child's mother or from their father.[106] This is due to the presence of genomic imprinting in the region. Beckwith-Wiedemann syndrome is also associated with genomic imprinting, often caused by abnormalities in maternal genomic imprinting of a region on chromosome 11.
Rett syndrome is underlied by mutations in the MECP2 gene despite no large-scale changes in expression of MeCP2 being found in microarray analyses. BDNF is downregulated in the MECP2 mutant resulting in Rett syndrome.
Transgenerational epigenetic observations
In the Överkalix study, Marcus Pembrey and colleagues observed that the paternal (but not maternal) grandsons[107] of Swedish men who were exposed during preadolescence to famine in the 19th century were less likely to die of cardiovascular disease. If food was plentiful, then diabetes mortality in the grandchildren increased, suggesting that this was a transgenerational epigenetic inheritance.[108] The opposite effect was observed for females—the paternal (but not maternal) granddaughters of women who experienced famine while in the womb (and therefore while their eggs were being formed) lived shorter lives on average.[109]
Cancer and developmental abnormalities
A variety of compounds are considered as epigenetic carcinogens—they result in an increased incidence of tumors, but they do not show mutagen activity (toxic compounds or pathogens that cause tumors incident to increased regeneration should also be excluded). Examples include diethylstilbestrol, arsenite, hexachlorobenzene, and nickel compounds.
Many teratogens exert specific effects on the fetus by epigenetic mechanisms.[110][111] While epigenetic effects may preserve the effect of a teratogen such as diethylstilbestrol throughout the life of an affected child, the possibility of birth defects resulting from exposure of fathers or in second and succeeding generations of offspring has generally been rejected on theoretical grounds and for lack of evidence.[112] However, a range of male-mediated abnormalities have been demonstrated, and more are likely to exist.[113] FDA label information for Vidaza, a formulation of 5-azacitidine (an unmethylatable analog of cytidine that causes hypomethylation when incorporated into DNA) states that "men should be advised not to father a child" while using the drug, citing evidence in treated male mice of reduced fertility, increased embryo loss, and abnormal embryo development.[114] In rats, endocrine differences were observed in offspring of males exposed to morphine.[115] In mice, second generation effects of diethylstilbesterol have been described occurring by epigenetic mechanisms.[116]
Recent studies have shown that the mixed-lineage leukemia (MLL) gene causes leukemia by rearranging and fusing with other genes in different chromosomes, which is a process under epigenetic control.[117]
Other investigations have concluded that alterations in histone acetylation and DNA methylation occur in various genes influencing prostate cancer.[118] Gene expression in the prostate can be modulated by nutrition and lifestyle changes.[119]
In 2008, the National Institutes of Health announced that $190 million had been earmarked for epigenetics research over the next five years. In announcing the funding, government officials noted that epigenetics has the potential to explain mechanisms of aging, human development, and the origins of cancer, heart disease, mental illness, as well as several other conditions. Some investigators, like Randy Jirtle, PhD, of Duke University Medical Center, think epigenetics may ultimately turn out to have a greater role in disease than genetics.[120]
DNA methylation in cancer
DNA methylation is an important regulator of gene transcription and a large body of evidence has demonstrated that aberrant DNA methylation is associated with unscheduled gene silencing, and the genes with high levels of 5-methylcytosine in their promoter region are transcriptionally silent. DNA methylation is essential during embryonic development, and in somatic cells, patterns of DNA methylation are in general transmitted to daughter cells with a high fidelity. Aberrant DNA methylation patterns have been associated with a large number of human malignancies and found in two distinct forms: hypermethylation and hypomethylation compared to normal tissue. Hypermethylation is one of the major epigenetic modifications that repress transcription via promoter region of tumour suppressor genes. Hypermethylation typically occurs at CpG islands in the promoter region and is associated with gene inactivation. Global hypomethylation has also been implicated in the development and progression of cancer through different mechanisms.[121]
DNA repair epigenetics in cancer
Germ line (familial) mutations have been identified in 34 different DNA repair genes that cause a high risk of cancer, including, for example BRCA1 and ATM. These are listed in the article DNA repair-deficiency disorder. However, cancers caused by such germ line mutations make up only a very small proportion of cancers. For instance, germ line mutations cause only 2% to 5% of colon cancer cases.[122]
Epigenetic reductions in expression of DNA repair genes, however, are very frequent in sporadic (non-germ line) cancers, as shown among some representative cancers in the table in this section, while mutations in DNA repair genes in sporadic cancer are very rare.[123]
| Cancer | Gene | Epigenetic change | Frequency | Ref. |
|---|---|---|---|---|
| Breast | BRCA1 | CpG island methylation | 13% | [124] |
| WRN | CpG island methylation | 17% | [125] | |
| Ovarian | WRN | CpG island methylation | 36% | [126] |
| BRCA1 | CpG island methylation | 5%–30% | [124][127][128] | |
| FANCF | CpG island methylation | 21% | [127] | |
| RAD51C | CpG island methylation | 3% | [127] | |
| Colorectal | MGMT | CpG island methylation | 40%–90% | [129][130][131][132][133] |
| WRN | CpG island methylation | 38% | [125] | |
| MLH1 | CpG island methylation | 2%–65% | [125][130][134] | |
| MSH2 | CpG island methylation | 13% | [131] | |
| ERCC1 | epigenetic type unknown | 100% | [135] | |
| Xpf | epigenetic type unknown | 55% | [135] | |
| Head and neck |
MGMT | CpG island methylation | 35%–57% | [136][137][138][139] |
| MLH1 | CpG island methylation | 27%–33% | [140][141][142] | |
| NEIL1 | CpG island methylation | 62% | [136] | |
| FANCB | CpG island methylation | 46% | [136] | |
| MSH4 | CpG island methylation | 46% | [136] | |
| ATM | CpG island methylation | 25% | [143] |
Deficiencies in expression of DNA repair genes cause increased mutation rates. Mutations rates increase in mice defective for mismatch DNA repair genes PMS2, MLH1, MSH2, MSH3 or MSH6[144][145] or for DNA repair gene BRCA2,[146] while chromosomal rearrangements and aneuploidy are noted to increase in humans defective in DNA repair gene BLM.[147] Thus, deficiency in DNA repair causes genome instability and this genome instability is likely the main underlying cause of the genetic alterations leading to cancer. In fact, the first event in many sporadic neoplasias is a heritable alteration that affects genetic instability and epigenetic defects in DNA repair are somatically heritable.[148]
Variant histones H2A in cancer
The histone variants of the H2A family are highly conserved in mammals, playing critical roles in regulating many nuclear processes by altering chromatin structure. One of the key H2A variants, H2A.X, marks DNA damage, facilitating the recruitment of DNA repair proteins to restore genomic integrity. Another variant, H2A.Z, plays an important role in both gene activation and repression. A high level of H2A.Z expression is ubiquitously detected in many cancers and is significantly associated with cellular proliferation and genomic instability.[121] Histone variant macroH2A1 is important in the pathogenesis of many types of cancers, for instance in hepatocellular carcinoma.[149]
Cancer treatment
Current research has shown that epigenetic pharmaceuticals could be a replacement or adjuvant therapy for currently accepted treatment methods such as radiation and chemotherapy, or could enhance the effects of these current treatments.[150] It has been shown that the epigenetic control of the proto-onco regions and the tumor suppressor sequences by conformational changes in histones directly affects the formation and progression of cancer.[151] Epigenetics also has the factor of reversibility, a characteristic that other cancer treatments do not offer.[118]
Drug development has focused mainly on histone acetyltransferase (HAT) and histone deacetylase (HDAC), and has included the introduction to the market of the new pharmaceutical vorinostat, an HDAC inhibitor.[152] HDAC has been shown to play an integral role in the progression of oral squamous cancer.[151]
Current front-runner candidates for new drug targets are histone lysine methyltransferases (KMT) and protein arginine methyltransferases (PRMT).[153]
Twin studies
Recent studies involving both dizygotic (fraternal) and monozygotic (identical) twins have produced some evidence of epigenetic influence in humans.[101][154][155]
Direct comparisons between identical twins constitute the ideal experimental model for testing environmental epigenetics, because DNA sequence differences that would be abundant in a singleton-based study do not interfere with the analysis. Research has shown that a difference in the environment can produce long-term epigenetic effects, and different developmental monozygotic twin subtypes may be different with respect to their susceptibility to be discordant from an epigenetic point of view.[156]
One of the first high-throughput studies of epigenetic differences between monozygotic twins focused in comparing global and locus-specific changes in DNA methylation and histone modifications in a sample of 40 monozygotic twin pairs.[101] In this case, only healthy twin pairs were studied, but a wide range of ages was represented, between 3 and 74 years. One of the major conclusions from this study was that there is an age-dependent accumulation of epigenetic differences between the two siblings of twin pairs. This accumulation suggests the existence of epigenetic “drift”.
A more recent study, where 114 monozygotic twins and 80 dizygotic twins were analyzed for the DNA methylation status of around 6000 unique genomic regions, concluded that epigenetic similarity at the time of blastocyst splitting may also contribute to phenotypic similarities in monozygotic co-twins. This supports the notion that microenvironment at early stages of embryonic development can be quite important for the establishment of epigenetic marks.[157]
Epigenetics in microorganisms

Bacteria make widespread use of postreplicative DNA methylation for the epigenetic control of DNA-protein interactions. Bacteria make use of DNA adenine methylation (rather than DNA cytosine methylation) as an epigenetic signal. DNA adenine methylation is important in bacteria virulence in organisms such as Escherichia coli, Salmonella, Vibrio, Yersinia, Haemophilus, and Brucella. In Alphaproteobacteria, methylation of adenine regulates the cell cycle and couples gene transcription to DNA replication. In Gammaproteobacteria, adenine methylation provides signals for DNA replication, chromosome segregation, mismatch repair, packaging of bacteriophage, transposase activity and regulation of gene expression.[158][159]
The filamentous fungus Neurospora crassa is a prominent model system for understanding the control and function of cytosine methylation. In this organisms, DNA methylation is associated with relics of a genome defense system called RIP (repeat-induced point mutation) and silences gene expression by inhibiting transcription elongation.[160]
The yeast prion PSI is generated by a conformational change of a translation termination factor, which is then inherited by daughter cells. This can provide a survival advantage under adverse conditions. This is an example of epigenetic regulation enabling unicellular organisms to respond rapidly to environmental stress. Prions can be viewed as epigenetic agents capable of inducing a phenotypic change without modification of the genome.[159]
Direct detection of epigenetic marks in microorganisms is possible with single molecule real time sequencing, in which polymerase sensitivity allows for measuring methylation and other modifications as a DNA molecule is being sequenced.[161] Several projects have demonstrated the ability to collect genome-wide epigenetic data in bacteria.[162][163][164][165]
See also
- B chromosome
- Baldwin effect
- Behavioral epigenetics
- Centromere
- Computational epigenetics
- Dutch famine of 1944 (Legacy)
- Epigenetic clock
- Degeneracy (biology)
- Ecotype
- Emergenesis
- Epigenetic Therapy
- Epigenomics
- Evolutionary capacitance
- Evolutionary developmental psychology
- Extranuclear inheritance
- Hologenome theory of evolution
- Human genome
- Molecular biology
- Molecular epidemiology
- Molecular pathological epidemiology
- Molecular pathology
- Nutriepigenomics
- Position-effect variegation
- Preformationism
- Somatic epitype
- Synthetic genetic array
- Weismann barrier
References
- ↑ 1.0 1.1 Ledford H (2008). "Disputed definitions". Nature 455 (7216): 1023–8. doi:10.1038/4551023a. PMID 18948925.
- ↑ Spector, Tim (2012). Identically Different: Why You Can Change Your Genes. London: Weidenfeld & Nicolson. p. 8.
Just over ten years ago researchers found that the diets of pregnant mothers could alter the behaviour of genes in their children and that these changes could last a lifetime and then be passed on in turn to their children. The genes were literally being switched on or off by a new mechanism we call epigenetics – meaning in Greek 'around the gene'. Contrary to traditional genetic dogma, these changes could be transferred to the next generation. In this case the mothers just happened to be rats, but recent similar findings in humans have created a revolution in our thinking.
- ↑ Carey N. (2011): Epigenetics revolution: How modern biology is rewriting our understanding of genetics, disease and inheritance. Icon Books, London, ISBN 978-1-84831-315-6; ISBN 978-1-84831-316-3.
- ↑ 4.0 4.1 Bird A (May 2007). "Perceptions of epigenetics". Nature 447 (7143): 396–8. Bibcode:2007Natur.447..396B. doi:10.1038/nature05913. PMID 17522671.
- ↑ "Special report: 'What genes remember' by Philip Hunter | Prospect Magazine May 2008 issue 146". Web.archive.org. 1 May 2008. Retrieved 26 July 2012.
- ↑ Reik W (May 2007). "Stability and flexibility of epigenetic gene regulation in mammalian development". Nature 447 (7143): 425–32. Bibcode:2007Natur.447..425R. doi:10.1038/nature05918. PMID 17522676.
- ↑ Waddington CH (1942). "The epigenotype". Endeavour 1: 18–20.
- ↑ According to the Oxford English Dictionary:
The word is used by W. Harvey, Exercitationes 1651, p. 148, and in the English Anatomical Exercitations 1653, p. 272. It is explained to mean ‘partium super-exorientium additamentum’, ‘the additament of parts budding one out of another’.
It is also worth quoting this adumbration of the definition given there (viz., "The formation of an organic germ as a new product"):theory of epigenesis: the theory that the germ is brought into existence (by successive accretions), and not merely developed, in the process of reproduction. [...] The opposite theory was formerly known as the ‘theory of evolution’; to avoid the ambiguity of this name, it is now spoken of chiefly as the ‘theory of preformation’, sometimes as that of ‘encasement’ or ‘emboîtement’.
- ↑ Waddington, Conrad H. 1953. The Epigenetics of birds. Cambridge University Press
- ↑ Hall BK (15 January 2004). "In search of evolutionary developmental mechanisms: the 30-year gap between 1944 and 1974". 302 (1): 5–18. doi:10.1002/jez.b.20002. PMID 14760651.
- ↑ Alvarez-Buylla ER, Chaos A, Aldana M, Benítez M, Cortes-Poza Y, Espinosa-Soto C et al. (November 3, 2008). "Floral Morphogenesis: Stochastic Explorations of a Gene Network Epigenetic Landscape.". PLoS ONE. doi:10.1371/journal.pone.0003626. PMID 18978941.
- ↑ 12.0 12.1 Rabajante JF, Babierra AL (January 30, 2015). "Branching and oscillations in the epigenetic landscape of cell-fate determination". Progress in Biophysics and Molecular Biology. doi:10.1016/j.pbiomolbio.2015.01.006. PMID 25641423.
- ↑ Gottlieb G (1991). "Epigenetic systems view of human development". Developmental Psychology 27 (1): 33–34. doi:10.1037/0012-1649.27.1.33.
- ↑ Gilbert Gottlieb. Probabilistic epigenesis, Developmental Science 10:1 (2007), 1–11
- ↑ Boeree, C. George, (1997/2006), Personality Theories, Erik Erikson
- ↑ Erikson, Erik (1968). Identity: Youth and Crisis. Chapter 3: W.W. Norton and Company, Inc. p. 92.
- ↑ "Epigenetics". Bio-Medicine.org. Retrieved 21 May 2011.
- ↑ Holliday R (Jan 30, 1990). "DNA Methylation and Epigenetic Inheritance". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 326 (1235): 329–338. doi:10.1098/rstb.1990.0015.
- ↑ 19.0 19.1 Riggs AD, Russo VEA, Martienssen RA (1996). Epigenetic mechanisms of gene regulation. Plainview, N.Y.: Cold Spring Harbor Laboratory Press. ISBN 0-87969-490-4.
- ↑ 20.0 20.1 "Overview". NIH Roadmap Epigenomics Project.
- ↑ Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A (2009). "An operational definition of epigenetics". Genes Dev. 23 (7): 781–3. doi:10.1101/gad.1787609. PMC 3959995. PMID 19339683.
- ↑ Chandler VL (February 2007). "Paramutation: from maize to mice". Cell 128 (4): 641–5. doi:10.1016/j.cell.2007.02.007. PMID 17320501.
- ↑ Kovalchuk O, Baulch JE (January 2008). "Epigenetic changes and nontargeted radiation effects—is there a link?". Environ. Mol. Mutagen. 49 (1): 16–25. doi:10.1002/em.20361. PMID 18172877.
- ↑ Ilnytskyy Y, Kovalchuk O (September 2011). "Non-targeted radiation effects-an epigenetic connection". Mutat. Res. 714 (1–2): 113–25. doi:10.1016/j.mrfmmm.2011.06.014. PMID 21784089.
- ↑ Friedl AA, Mazurek B, Seiler DM (2012). "Radiation-induced alterations in histone modification patterns and their potential impact on short-term radiation effects". Front Oncol 2: 117. doi:10.3389/fonc.2012.00117. PMC 3445916. PMID 23050241.
- ↑ Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A et al. (July 2007). "DNA damage, homology-directed repair, and DNA methylation". PLoS Genet. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- ↑ O'Hagan HM, Mohammad HP, Baylin SB (2008). Lee JT, ed. "Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island". PLoS Genet. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- ↑ Malanga M, Althaus FR (2005). "The role of poly(ADP-ribose) in the DNA damage signaling network". Biochem Cell Biol 83 (3): 354–364. doi:10.1139/o05-038. PMID 15959561.
- ↑ Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK et al. (August 2009). "Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler". Proc. Natl. Acad. Sci. U.S.A. 106 (33): 13770–4. Bibcode:2009PNAS..10613770G. doi:10.1073/pnas.0906920106. PMC 2722505. PMID 19666485.
- ↑ Lin JC, Jeong S, Liang G, Takai D, Fatemi M, Tsai YC et al. (November 2007). "Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island". Cancer Cell 12 (5): 432–44. doi:10.1016/j.ccr.2007.10.014. PMID 17996647.
- ↑ Tabish AM, Poels K, Hoet P, Godderis L (2012). Chiariotti L, ed. "Epigenetic factors in cancer risk: effect of chemical carcinogens on global DNA methylation pattern in human TK6 cells". PLoS ONE 7 (4): e34674. Bibcode:2012PLoSO...734674T. doi:10.1371/journal.pone.0034674. PMC 3324488. PMID 22509344.
- ↑ Burdge GC, Hoile SP, Uller T, Thomas NA, Gluckman PD, Hanson MA et al. (2011). Imhof A, ed. "Progressive, transgenerational changes in offspring phenotype and epigenotype following nutritional transition". PLoS ONE 6 (11): e28282. Bibcode:2011PLoSO...628282B. doi:10.1371/journal.pone.0028282. PMC 3227644. PMID 22140567.
- ↑ Fang M, Chen D, Yang CS (January 2007). "Dietary polyphenols may affect DNA methylation". J. Nutr. 137 (1 Suppl): 223S–228S. PMID 17182830.
- ↑ Olaharski AJ, Rine J, Marshall BL, Babiarz J, Zhang L, Verdin E et al. (December 2005). "The flavoring agent dihydrocoumarin reverses epigenetic silencing and inhibits sirtuin deacetylases". PLoS Genet. 1 (6): e77. doi:10.1371/journal.pgen.0010077. PMC 1315280. PMID 16362078.
- ↑ Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y et al. (August 2008). "Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells". Int. J. Cancer 123 (3): 552–60. doi:10.1002/ijc.23590. PMID 18431742.
- ↑ Davis JN, Kucuk O, Djuric Z, Sarkar FH (June 2001). "Soy isoflavone supplementation in healthy men prevents NF-kappa B activation by TNF-alpha in blood lymphocytes". Free Radic. Biol. Med. 30 (11): 1293–302. doi:10.1016/S0891-5849(01)00535-4. PMID 11368927.
- ↑ Djuric Z, Chen G, Doerge DR, Heilbrun LK, Kucuk O (October 2001). "Effect of soy isoflavone supplementation on markers of oxidative stress in men and women". Cancer Lett. 172 (1): 1–6. doi:10.1016/S0304-3835(01)00627-9. PMID 11595123.
- ↑ Kropat C, Mueller D, Boettler U, Zimmermann K, Heiss EH, Dirsch VM et al. (March 2013). "Modulation of Nrf2-dependent gene transcription by bilberry anthocyanins in vivo". Mol Nutr Food Res 57 (3): 545–50. doi:10.1002/mnfr.201200504. PMID 23349102.
- ↑ Baron R, Vellore NA (2012). "LSD1/CoREST is an allosteric nanoscale clamp regulated by H3-histone-tail molecular recognition". Proc Natl Acad Sci U S A. 109 (31): 12509–14. Bibcode:2012PNAS..10912509B. doi:10.1073/pnas.1207892109. PMC 3411975. PMID 22802671.
- ↑ Jablonka E, Lamb MJ, Lachmann M (September 1992). "Evidence, mechanisms and models for the inheritance of acquired characteristics". J. Theor. Biol. 158 (2): 245–268. doi:10.1016/S0022-5193(05)80722-2.
- ↑ Ptashne M (April 2007). "On the use of the word 'epigenetic'". Curr. Biol. 17 (7): R233–6. doi:10.1016/j.cub.2007.02.030. PMID 17407749.
- ↑ Jenuwein T, Laible G, Dorn R, Reuter G (January 1998). "SET domain proteins modulate chromatin domains in eu- and heterochromatin". Cell. Mol. Life Sci. 54 (1): 80–93. doi:10.1007/s000180050127. PMID 9487389.
- ↑ Slotkin RK, Martienssen R (April 2007). "Transposable elements and the epigenetic regulation of the genome". Nature Reviews Genetics 8 (4): 272–85. doi:10.1038/nrg2072. PMID 17363976.
- ↑ 44.0 44.1 Li E, Bestor TH, Jaenisch R (June 1992). "Targeted mutation of the DNA methyltransferase gene results in embryonic lethality". Cell 69 (6): 915–26. doi:10.1016/0092-8674(92)90611-F. PMID 1606615.
- ↑ Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA et al. (June 1999). "The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors". Nucleic Acids Res. 27 (11): 2291–8. doi:10.1093/nar/27.11.2291. PMC 148793. PMID 10325416.
- ↑ Leonhardt H, Page AW, Weier HU, Bestor TH (November 1992). "A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei". Cell 71 (5): 865–73. doi:10.1016/0092-8674(92)90561-P. PMID 1423634.
- ↑ Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF (September 1997). "Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1". Science 277 (5334): 1996–2000. doi:10.1126/science.277.5334.1996. PMID 9302295.
- ↑ Robertson KD, Wolffe AP (October 2000). "DNA methylation in health and disease". Nature Reviews Genetics 1 (1): 11–9. doi:10.1038/35049533. PMID 11262868.
- ↑ Li E, Beard C, Jaenisch R (November 1993). "Role for DNA methylation in genomic imprinting". Nature 366 (6453): 362–5. Bibcode:1993Natur.366..362L. doi:10.1038/366362a0. PMID 8247133.
- ↑ Viens A, Mechold U, Brouillard F, Gilbert C, Leclerc P, Ogryzko V (July 2006). "Analysis of human histone H2AZ deposition in vivo argues against its direct role in epigenetic templating mechanisms". Mol. Cell. Biol. 26 (14): 5325–35. doi:10.1128/MCB.00584-06. PMC 1592707. PMID 16809769.
- ↑ Ogryzko VV (2008). "Erwin Schroedinger, Francis Crick and epigenetic stability". Biol. Direct 3: 15. doi:10.1186/1745-6150-3-15. PMC 2413215. PMID 18419815.
- ↑ Nottke A, Colaiácovo MP, Shi Y (March 2009). "Developmental roles of the histone lysine demethylases". Development 136 (6): 879–89. doi:10.1242/dev.020966. PMC 2692332. PMID 19234061.
- ↑ Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ (2009). "Determination of enriched histone modifications in non-genic portions of the human genome". BMC Genomics 10: 143. doi:10.1186/1471-2164-10-143. PMC 2667539. PMID 19335899.
- ↑ Sneppen K, Micheelsen MA, Dodd IB (April 15, 2008). "Ultrasensitive gene regulation by positive feedback loops in nucleosome modification". Molecular systems biology 4 (1): 182. doi:10.1038/msb.2008.21. PMC 2387233. PMID 18414483. Retrieved 5 May 2014.
- ↑ "Epigenetic cell memory". Cmol.nbi.dk. Retrieved 26 July 2012.
- ↑ Dodd IB, Micheelsen MA, Sneppen K, Thon G (May 2007). "Theoretical analysis of epigenetic cell memory by nucleosome modification". Cell 129 (4): 813–22. doi:10.1016/j.cell.2007.02.053. PMID 17512413.
- ↑ Morris KL (2008). "Epigenetic Regulation of Gene Expression". RNA and the Regulation of Gene Expression: A Hidden Layer of Complexity. Norfolk, England: Caister Academic Press. ISBN 1-904455-25-5.
- ↑ Mattick JS, Amaral PP, Dinger ME, Mercer TR, Mehler MF (January 2009). "RNA regulation of epigenetic processes". BioEssays 31 (1): 51–9. doi:10.1002/bies.080099. PMID 19154003.
- ↑ Choi CQ (25 May 2006). "The Scientist: RNA can be hereditary molecule". The Scientist. Retrieved 2006.
- ↑ 60.0 60.1 60.2 Bernal JE, Duran C, Papiha SS (2012). "Transcriptional and epigenetic regulation of human microRNAs". Cancer Lett 331 (1): 1–10. doi:10.1016/j.canlet.2012.12.006. PMID 3246373.
- ↑ Browse miRBase by species
- ↑ Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J et al. (2005). "Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs". Nature 433 (7027): 769–773. doi:10.1038/nature03315. PMID 15685193.
- ↑ Lee D, Shin C (2012). MicroRNA-target interactions: new insights from genome-wide approaches" Ann N Y Acad Sci 1271:118-28. doi: 10.1111/j.1749-6632.2012.06745.x. Review. PMID 23050973
- ↑ Friedman RC, Farh KK, Burge CB, Bartel DP (2009). "Most mammalian mRNAs are conserved targets of microRNAs". Genome Res 19 (1): 92–105. doi:10.1101/gr.082701.108. PMC 2612969. PMID 18955434.
- ↑ Goll MG, Bestor TH (2005). "Eukaryotic cytosine methyltransferases". Annu Rev Biochem 74: 481–514. doi:10.1146/annurev.biochem.74.010904.153721. PMID 15952895.
- ↑ Guifang Jia; Ye Fu; Xu Zhao; Qing Dai; Guanqun Zheng; Ying Yang; Chengqi Yi; Lindahl, Tomas; Tao Pan; Yun-Gui Yang; Chuan He (16 October 2011). "N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO". Nature Chemical Biology 7 (12): 885–887. doi:10.1038/nchembio.687. PMC 3218240. PMID 22002720.
- ↑ "New research links common RNA modification to obesity". Physorg.com. Retrieved 26 July 2012.
- ↑ Howden BP, Beaume M, Harrison PF, Hernandez D, Schrenzel J, Seemann T et al. (August 2013). "Analysis of the Small RNA Transcriptional Response in Multidrug-Resistant Staphylococcus aureus after Antimicrobial Exposure". Antimicrob. Agents Chemother. 57 (8): 3864–74. doi:10.1128/AAC.00263-13. PMC 3719707. PMID 23733475.
- ↑ sRNATarBase 2.0 A comprehensive database of bacterial SRNA targets verified by experiments
- ↑ Genomics maps for small non-coding RNA's and their targets in microbial genomes
- ↑ Yool A, Edmunds WJ (1998). "Epigenetic inheritance and prions". Journal of Evolutionary Biology 11 (2): 241–242. doi:10.1007/s000360050085.
- ↑ Cox BS (1965). "[PSI], a cytoplasmic suppressor of super-suppression in yeast". Heredity 20 (4): 505–521. doi:10.1038/hdy.1965.65.
- ↑ Lacroute F (May 1971). "Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast". J. Bacteriol. 106 (2): 519–22. PMC 285125. PMID 5573734.
- ↑ Liebman SW, Sherman F (September 1979). "Extrachromosomal psi+ determinant suppresses nonsense mutations in yeast". J. Bacteriol. 139 (3): 1068–71. PMC 218059. PMID 225301.
- ↑ True HL, Lindquist SL (September 2000). "A yeast prion provides a mechanism for genetic variation and phenotypic diversity". Nature 407 (6803): 477–83. doi:10.1038/35035005. PMID 11028992.
- ↑ Shorter J, Lindquist S (June 2005). "Prions as adaptive conduits of memory and inheritance". Nature Reviews Genetics 6 (6): 435–50. doi:10.1038/nrg1616. PMID 15931169.
- ↑ Giacomelli MG, Hancock AS, Masel J (2007). "The conversion of 3′ UTRs into coding regions". Molecular Biology & Evolution 24 (2): 457–464. doi:10.1093/molbev/msl172. PMC 1808353. PMID 17099057.
- ↑ Lancaster AK, Bardill JP, True HL, Masel J (2010). "The Spontaneous Appearance Rate of the Yeast Prion PSI+ and Its Implications for the Evolution of the Evolvability Properties of the PSI+ System". Genetics 184 (2): 393–400. doi:10.1534/genetics.109.110213. PMC 2828720. PMID 19917766.
- ↑ Sapp J (1991). "Concepts of organization. The leverage of ciliate protozoa". Dev. Biol. (NY) 7: 229–58. doi:10.1007/978-1-4615-6823-0_11. PMID 1804215.
- ↑ Sapp J (2003). Genesis: the evolution of biology. Oxford [Oxfordshire]: Oxford University Press. ISBN 0-19-515619-6.
- ↑ Gray RD, Oyama S, Griffiths PE (2003). Cycles of Contingency: Developmental Systems and Evolution (Life and Mind: Philosophical Issues in Biology and Psychology). Cambridge, Mass: The MIT Press. ISBN 0-262-65063-0.
- ↑ Chapter: "Nervous System Development" in "Epigenetics," by Benedikt Hallgrimsson and Brian Hall
- ↑ Costa S, Shaw P (March 2007). "'Open minded' cells: how cells can change fate" (PDF). Trends Cell Biol. 17 (3): 101–6. doi:10.1016/j.tcb.2006.12.005. PMID 17194589.
This might suggest that plant cells do not use or require a cellular memory mechanism and just respond to positional information. However, it has been shown that plants do use cellular memory mechanisms mediated by PcG proteins in several processes, ... (p.104)
- ↑ Griesemer J, Haber MH, Yamashita G, Gannett L (March 2005). "Critical Notice: Cycles of Contingency – Developmental Systems and Evolution". Biology & Philosophy 20 (2–3): 517–544. doi:10.1007/s10539-004-0836-4.
- ↑ Chahwan R, Wontakal SN, Roa S (March 2011). "The multidimensional nature of epigenetic information and its role in disease". Discov Med 11 (58): 233–43. PMID 21447282.
- ↑ Online 'Mendelian Inheritance in Man' (OMIM) 105830
- ↑ Lamb MJ, Jablonka E (2005). Evolution in four dimensions: genetic, epigenetic, behavioral, and symbolic variation in the history of life. Cambridge, Mass: MIT Press. ISBN 0-262-10107-6.
- ↑ See also Denis Noble The Music of Life see esp pp. 93–8 and p. 48 where he cites Jablonka & Lamb and Massimo Pigliucci's review of Jablonka and Lamb in Nature 435, 565–566 (2 June 2005)
- ↑ Maynard Smith J (1990). "Models of a Dual Inheritance System". Journal of Theoretical Biology 143 (1): 41–53. doi:10.1016/S0022-5193(05)80287-5. PMID 2359317.
- ↑ Lynch M (2007). "The frailty of adaptive hypotheses for the origins of organismal complexity". PNAS 104 (suppl. 1): 8597–8604. Bibcode:2007PNAS..104.8597L. doi:10.1073/pnas.0702207104. PMC 1876435. PMID 17494740.
- ↑ Rando OJ, Verstrepen KJ (February 2007). "Timescales of genetic and epigenetic inheritance". Cell 128 (4): 655–68. doi:10.1016/j.cell.2007.01.023. PMID 17320504.
- ↑ Lancaster AK, Masel J (1 September 2009). "The evolution of reversible switches in the presence of irreversible mimics". Evolution 63 (9): 2350–2362. doi:10.1111/j.1558-5646.2009.00729.x. PMC 2770902. PMID 19486147.
- ↑ Griswold CK, Masel J (2009). Úbeda F, ed. "Complex Adaptations Can Drive the Evolution of the Capacitor PSI+, Even with Realistic Rates of Yeast Sex". PLoS Genetics 5 (6): e1000517. doi:10.1371/journal.pgen.1000517. PMC 2686163. PMID 19521499.
- ↑ Cooney CA, Dave AA, Wolff GL (August 2002). "Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring". J. Nutr. 132 (8 Suppl): 2393S–2400S. PMID 12163699.
- ↑ Waterland RA, Jirtle RL (August 2003). "Transposable elements: targets for early nutritional effects on epigenetic gene regulation". Mol. Cell. Biol. 23 (15): 5293–300. doi:10.1128/MCB.23.15.5293-5300.2003. PMC 165709. PMID 12861015.
- ↑ Fearful Memories Passed Down to Mouse Descendants: Genetic imprint from traumatic experiences carries through at least two generations, By Ewen Callaway and Nature magazine | Sunday, 1 December 2013.
- ↑ Mice can 'warn' sons, grandsons of dangers via sperm, by Mariette Le Roux, 12/1/13.
- ↑ G. Francis, "Too Much Success for Recent Groundbreaking Epigenetic Experiments" http://www.genetics.org/content/198/2/449.abstract
- ↑ http://www.ncbi.nlm.nih.gov/pubmed/24292232 (see comment by Gonzalo Otazu)
- ↑ http://www.the-scientist.com/?articles.view/articleNo/41239/title/Epigenetics-Paper-Raises-Questions/
- ↑ 101.0 101.1 101.2 Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML et al. (July 2005). "Epigenetic differences arise during the lifetime of monozygotic twins". Proc. Natl. Acad. Sci. U.S.A. 102 (30): 10604–9. Bibcode:2005PNAS..10210604F. doi:10.1073/pnas.0500398102. PMC 1174919. PMID 16009939.
- ↑ Jablonka E, Raz G (June 2009). "Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution". Q Rev Biol 84 (2): 131–76. doi:10.1086/598822. PMID 19606595.
- ↑ Davies, Hazel (2008). Do Butterflies Bite?: Fascinating Answers to Questions about Butterflies and Moths (Animals Q&A). Rutgers University Press.
- ↑ Chahwan R, Wontakal SN, Roa S (October 2010). "Crosstalk between genetic and epigenetic information through cytosine deamination". Trends Genet. 26 (10): 443–8. doi:10.1016/j.tig.2010.07.005. PMID 20800313.
- ↑ Wood AJ, Oakey RJ (November 2006). "Genomic imprinting in mammals: emerging themes and established theories". PLoS Genet. 2 (11): e147. doi:10.1371/journal.pgen.0020147. PMC 1657038. PMID 17121465.
- ↑ Knoll JH, Nicholls RD, Magenis RE, Graham JM, Lalande M, Latt SA (February 1989). "Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion". Am. J. Med. Genet. 32 (2): 285–90. doi:10.1002/ajmg.1320320235. PMID 2564739.
- ↑ A person's paternal grandson is the son of a son of that person; a maternal grandson is the son of a daughter.
- ↑ Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjöström M et al. (February 2006). "Sex-specific, male-line transgenerational responses in humans". Eur. J. Hum. Genet. 14 (2): 159–66. doi:10.1038/sj.ejhg.5201538. PMID 16391557. Robert Winston refers to this study in a lecture; see also discussion at Leeds University, here
- ↑ "NOVA | Transcripts | Ghost in Your Genes". PBS. 16 October 2007. Retrieved 26 July 2012.
- ↑ Bishop JB, Witt KL, Sloane RA (December 1997). "Genetic toxicities of human teratogens". Mutat. Res. 396 (1–2): 9–43. doi:10.1016/S0027-5107(97)00173-5. PMID 9434858.
- ↑ Gurvich N, Berman MG, Wittner BS, Gentleman RC, Klein PS, Green JB (July 2005). "Association of valproate-induced teratogenesis with histone deacetylase inhibition in vivo". FASEB J. 19 (9): 1166–8. doi:10.1096/fj.04-3425fje. PMID 15901671.
- ↑ Smithells D (November 1998). "Does thalidomide cause second generation birth defects?". Drug Saf 19 (5): 339–41. doi:10.2165/00002018-199819050-00001. PMID 9825947.
- ↑ Friedler G (December 1996). "Paternal exposures: impact on reproductive and developmental outcome. An overview". Pharmacol. Biochem. Behav. 55 (4): 691–700. doi:10.1016/S0091-3057(96)00286-9. PMID 8981601.
- ↑ WebCite query result
- ↑ Cicero TJ, Adams ML, Giordano A, Miller BT, O'Connor L, Nock B (March 1991). "Influence of morphine exposure during adolescence on the sexual maturation of male rats and the development of their offspring". J. Pharmacol. Exp. Ther. 256 (3): 1086–93. PMID 2005573.
- ↑ Newbold RR, Padilla-Banks E, Jefferson WN (June 2006). "Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations". Endocrinology 147 (6 Suppl): S11–7. doi:10.1210/en.2005-1164. PMID 16690809.
- ↑ Mandal SS (April 2010). "Mixed lineage leukemia: versatile player in epigenetics and human disease". FEBS J. 277 (8): 1789. doi:10.1111/j.1742-4658.2010.07605.x. PMID 20236314.
- ↑ 118.0 118.1 Li LC, Carroll PR, Dahiya R (January 2005). "Epigenetic changes in prostate cancer: implication for diagnosis and treatment". J. Natl. Cancer Inst. 97 (2): 103–15. doi:10.1093/jnci/dji010. PMID 15657340.
- ↑ Ornish D, Magbanua MJ, Weidner G, Weinberg V, Kemp C, Green C et al. (June 2008). "Changes in prostate gene expression in men undergoing an intensive nutrition and lifestyle intervention". Proc. Natl. Acad. Sci. U.S.A. 105 (24): 8369–74. Bibcode:2008PNAS..105.8369O. doi:10.1073/pnas.0803080105. PMC 2430265. PMID 18559852.
- ↑ Beil, Laura (Winter 2008). "Medicine's New Epicenter? Epigenetics: New field of epigenetics may hold the secret to flipping cancer's "off" switch.". CURE (Cancer Updates, Research and Education).
- ↑ 121.0 121.1 Wong NC, Craig JM (2011). Epigenetics: A Reference Manual. Norfolk, England: Caister Academic Press. ISBN 1-904455-88-3.
- ↑ Jasperson KW, Tuohy TM, Neklason DW, Burt RW (2010). "Hereditary and familial colon cancer". Gastroenterology 138 (6): 2044–2058. doi:10.1053/j.gastro.2010.01.054. PMID 20420945.
- ↑ Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ et al. (2007). "The genomic landscapes of human breast and colorectal cancers". Science 318 (5853): 1108–1113. doi:10.1126/science.1145720. PMID 17932254.
- ↑ 124.0 124.1 Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E et al. (Apr 2000). "Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors.". J Natl Cancer Inst 92 (7): 564–9. doi:10.1093/jnci/92.7.564. PMID 10749912.
- ↑ 125.0 125.1 125.2 Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF et al. (Jun 2006). "Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer.". Proc Natl Acad Sci U S A 103 (23): 8822–7. doi:10.1073/pnas.0600645103. PMID 16723399.
- ↑ Baldwin RL, Nemeth E, Tran H, Shvartsman H, Cass I, Narod S et al. (2000). "BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study". Cancer Res 60 (19): 5329–5333. PMID 11034065.
- ↑ 127.0 127.1 127.2 Rigakos G, Razis E (2012). "BRCAness: finding the Achilles heel in ovarian cancer". Oncologist 17 (7): 956–62. doi:10.1634/theoncologist.2012-0028. PMC 3399652. PMID 22673632.
- ↑ Stefansson OA, Villanueva A, Vidal A, Martí L, Esteller M (2012). "BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer". Epigenetics 7 (11): 1225–1229. doi:10.4161/epi.22561. PMID 23069641.
- ↑ Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J et al. (2005). "MGMT promoter methylation and field defect in sporadic colorectal cancer". J Natl Cancer Inst 97 (18): 1330–1338. doi:10.1093/jnci/dji275. PMID 16174854.
- ↑ 130.0 130.1 Psofaki V, Kalogera C, Tzambouras N, Stephanou D, Tsianos E, Seferiadis K et al. (2010). "Promoter methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal adenomas". World J Gastroenterol 16 (28): 3553–3560. doi:10.3748/wjg.v16.i28.3553. PMID 20653064.
- ↑ 131.0 131.1 Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Park CS et al. (2011). "Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence". Langenbecks Arch Surg 396 (7): 1017–1026. doi:10.1007/s00423-011-0812-9. PMID 21706233.
- ↑ Amatu A, Sartore-Bianchi A, Moutinho C, Belotti A, Bencardino K, Chirico G, Cassingena A, Rusconi F, Esposito A, Nichelatti M, Esteller M, Siena S (2013). "Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer". Clin. Cancer Res. 19 (8): 2265–72. doi:10.1158/1078-0432.CCR-12-3518. PMID 23422094.
- ↑ Mokarram P, Zamani M, Kavousipour S, Naghibalhossaini F, Irajie C, Moradi Sarabi M, Hosseini SV (2013). "Different patterns of DNA methylation of the two distinct O6-methylguanine-DNA methyltransferase (O6-MGMT) promoter regions in colorectal cancer". Mol. Biol. Rep. 40 (5): 3851–7. doi:10.1007/s11033-012-2465-3. PMID 23271133.
- ↑ Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO et al. (2005). "Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer". Gastroenterology 128 (5): 1160–71. doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- ↑ 135.0 135.1 Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B et al. (2012). "Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer". Genome Integr 3 (1): 3. doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- ↑ 136.0 136.1 136.2 136.3 Chaisaingmongkol J, Popanda O, Warta R, Dyckhoff G, Herpel E, Geiselhart L et al. (2012). "Epigenetic screen of human DNA repair genes identifies aberrant promoter methylation of NEIL1 in head and neck squamous cell carcinoma". Oncogene 31 (49): 5108–16. doi:10.1038/onc.2011.660. PMID 22286769.
- ↑ Fan CY (Mar 2004). "Epigenetic alterations in head and neck cancer: prevalence, clinical significance, and implications.". Curr Oncol Rep 6 (2): 152–61. doi:10.1007/s11912-004-0027-0. PMID 14751093.
- ↑ Koutsimpelas D, Pongsapich W, Heinrich U, Mann S, Mann WJ, Brieger J (2012). "Promoter methylation of MGMT, MLH1 and RASSF1A tumor suppressor genes in head and neck squamous cell carcinoma: pharmacological genome demethylation reduces proliferation of head and neck squamous carcinoma cells". Oncol Rep 27 (4): 1135–41. doi:10.3892/or.2012.1624. PMID 22246327.
- ↑ Sun W, Zaboli D, Liu Y, Arnaoutakis D, Khan T, Wang H et al. (2012). "Comparison of promoter hypermethylation pattern in salivary rinses collected with and without an exfoliating brush from patients with HNSCC". PLOS ONE 7 (3): e33642. doi:10.1371/journal.pone.0033642. PMID 22438973.
- ↑ Puri SK, Si L, Fan CY, Hanna E. "Aberrant promoter hypermethylation of multiple genes in head and neck squamous cell carcinoma.". Am J Otolaryngol 26 (1): 12–7. doi:10.1016/j.amjoto.2004.06.007. PMID 15635575.
- ↑ Zuo C, Zhang H, Spencer HJ, Vural E, Suen JY, Schichman SA et al. (2009). "Increased microsatellite instability and epigenetic inactivation of the hMLH1 gene in head and neck squamous cell carcinoma". Otolaryngol Head Neck Surg 141 (4): 484–490. doi:10.1016/j.otohns.2009.07.007. PMID 19786217.
- ↑ Tawfik HM, El-Maqsoud NM, Hak BH, El-Sherbiny YM (2011). "Head and neck squamous cell carcinoma: mismatch repair immunohistochemistry and promoter hypermethylation of hMLH1 gene". Am J Otolaryngol 32 (6): 528–536. doi:10.1016/j.amjoto.2010.11.005. PMID 21353335.
- ↑ Ai L, Vo QN, Zuo C, Li L, Ling W, Suen JY et al. (Jan 2004). "Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: promoter hypermethylation with clinical correlation in 100 cases.". Cancer Epidemiol Biomarkers Prev 13 (1): 150–6. doi:10.1158/1055-9965.epi-082-3. PMID 14744748.
- ↑ Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (1997). "Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2". Proc Natl Acad Sci U S A 94 (7): 3122–3127. doi:10.1073/pnas.94.7.3122. PMC 20332. PMID 9096356.
- ↑ Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM (Dec 2006). "Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6". Carcinogenesis 27 (12): 2402–2408. doi:10.1093/carcin/bgl079. PMC 2612936. PMID 16728433.
- ↑ Tutt AN, van Oostrom CT, Ross GM, van Steeg H, Ashworth A (2002). "Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation". EMBO Rep. 3 (3): 255–60. doi:10.1093/embo-reports/kvf037. PMC 1084010. PMID 11850397.
- ↑ German J (Mar 1969). "Bloom's syndrome. I. Genetical and clinical observations in the first twenty-seven patients." 21 (2). pp. 196–227. PMC 1706430. PMID 5770175.
- ↑ Nowak MA, Komarova NL, Sengupta A, Jallepalli PV, Shih I, Vogelstein B et al. (2002). "The role of chromosomal instability in tumor initiation". Proc Natl Acad Sci U S A 99 (25): 16226–16231. doi:10.1073/pnas.202617399. PMC 138593. PMID 12446840.
- ↑ Rappa F, Greco A, Podrini C, Cappello F, Foti M, Bourgoin L et al. (2013). Folli F, ed. "Immunopositivity for histone macroH2A1 isoforms marks steatosis-associated hepatocellular carcinoma". PLoS ONE 8 (1): e54458. Bibcode:2013PLoSO...854458R. doi:10.1371/journal.pone.0054458. PMC 3553099. PMID 23372727.
- ↑ Wang LG, Chiao JW (September 2010). "Prostate cancer chemopreventive activity of phenethyl isothiocyanate through epigenetic regulation (review)". Int. J. Oncol. 37 (3): 533–9. doi:10.3892/ijo_00000702. PMID 20664922.
- ↑ 151.0 151.1 Iglesias-Linares A, Yañez-Vico RM, González-Moles MA (May 2010). "Potential role of HDAC inhibitors in cancer therapy: insights into oral squamous cell carcinoma". Oral Oncol. 46 (5): 323–9. doi:10.1016/j.oraloncology.2010.01.009. PMID 20207580.
- ↑ Spannhoff A, Sippl W, Jung M (January 2009). "Cancer treatment of the future: inhibitors of histone methyltransferases". Int. J. Biochem. Cell Biol. 41 (1): 4–11. doi:10.1016/j.biocel.2008.07.024. PMID 18773966.
- ↑ Dowden J, Hong W, Parry RV, Pike RA, Ward SG (April 2010). "Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases". Bioorg. Med. Chem. Lett. 20 (7): 2103–5. doi:10.1016/j.bmcl.2010.02.069. PMID 20219369.
- ↑ Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH et al. (February 2009). "DNA methylation profiles in monozygotic and dizygotic twins". Nat. Genet. 41 (2): 240–5. doi:10.1038/ng.286. PMID 19151718.
- ↑ O'Connor, Anahad (11 March 2008). "The Claim: Identical Twins Have Identical DNA". New York Times. Retrieved 2 May 2010.
- ↑ Ballestar E (2010). "Epigenetics lessons from twins: prospects for autoimmune disease". Clin Rev Allergy Immunol 39 (1): 30–41. doi:10.1007/s12016-009-8168-4. PMID 19653134.
- ↑ Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH et al. (2009). "DNA methylation profiles in monozygotic and dizygotic twins". Nat Genet 41 (2): 240–245. doi:10.1038/ng.286. PMID 19151718.
- ↑ Casadesús J, Low D (September 2006). "Epigenetic gene regulation in the bacterial world". Microbiol. Mol. Biol. Rev. 70 (3): 830–56. doi:10.1128/MMBR.00016-06. PMC 1594586. PMID 16959970.
- ↑ 159.0 159.1 Jorg Tost (2008). Epigenetics. Norfolk, England: Caister Academic Press. ISBN 1-904455-23-9.
- ↑ Lewis ZA, Honda S, Khlafallah TK, Jeffress JK, Freitag M, Mohn F et al. (March 2009). "Relics of repeat-induced point mutation direct heterochromatin formation in Neurospora crassa". Genome Res. 19 (3): 427–37. doi:10.1101/gr.086231.108. PMC 2661801. PMID 19092133.
- ↑ Schadt EE, Banerjee O, Fang G, Feng Z, Wong WH, Zhang X et al. (2012). "Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases". Genome Research 23 (1): 129–41. doi:10.1101/gr.136739.111. PMC 3530673. PMID 23093720.
- ↑ Davis BM, Chao MC, Waldor MK (2013). "Entering the era of bacterial epigenomics with single molecule real time DNA sequencing". Current Opinion in Microbiology 16 (2): 192–8. doi:10.1016/j.mib.2013.01.011. PMC 3646917. PMID 23434113.
- ↑ Lluch-Senar M, Luong K, Lloréns-Rico V, Delgado J, Fang G, Spittle K et al. (2013). Richardson PM, ed. "Comprehensive Methylome Characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at Single-Base Resolution". PLoS Genetics 9 (1): e1003191. doi:10.1371/journal.pgen.1003191. PMC 3536716. PMID 23300489.
- ↑ Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K et al. (2012). "The methylomes of six bacteria". Nucleic Acids Research 40 (22): 11450–62. doi:10.1093/nar/gks891. PMC 3526280. PMID 23034806.
- ↑ Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O et al. (2012). "Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing". Nature Biotechnology 30 (12): 1232–9. doi:10.1038/nbt.2432. PMID 23138224.
External links
- Haque FN, Gottesman II, Wong AH (May 2009). "Not really identical: epigenetic differences in monozygotic twins and implications for twin studies in psychiatry". Am J Med Genet C Semin Med Genet 151C (2): 136–41. doi:10.1002/ajmg.c.30206. PMID 19378334.
- The Human Epigenome Project (HEP)
- The Epigenome Network of Excellence (NoE)
- Canadian Epigenetics, Environment and Health Research Consortium (CEEHRC)
- The Epigenome Network of Excellence (NoE)- public international site
- DNA Is Not Destiny – Discover Magazine cover story
- BBC – Horizon – 2005 – The Ghost In Your Genes
- Epigenetics article at Hopkins Medicine
- Towards a global map of epigenetic variation
| ||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||