Electronic density
In quantum mechanics, and in particular quantum chemistry, the electronic density is a measure of the probability of an electron occupying an infinitesimal element of space surrounding any given point. It is a scalar quantity depending upon three spatial variables and is typically denoted as either ρ(r) or n(r). The density is determined, through definition, by the normalized N-electron wavefunction which itself depends upon 4N variables (3N spatial and N spin coordinates). Conversely, the density determines the wave function modulo a phase factor, providing the formal foundation of density functional theory.
Definition
The electronic density corresponding to a normalized N-electron wavefunction (with r and s denoting spatial and spin variables respectively) is defined as[1]
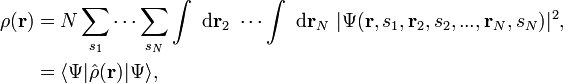
where the operator corresponding to the density observable is
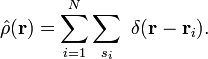
In Hartree–Fock and density functional theories the wave function is typically represented as a single Slater determinant constructed from N orbitals, φk, with corresponding occupations nk. In these situations the density simplifies to
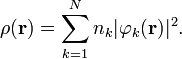
General Properties
From its definition, the electron density is a non-negative function integrating to the total number of electrons. Further, for a system with kinetic energy T, the density satisfies the inequalities[2]
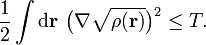
For finite kinetic energies, the first (stronger) inequality places the square root of the density in the Sobolev space H1(R3). Together with the normalization and non-negativity this defines a space containing physically acceptable densities as
The second inequality places the density in the L3 space. Together with the normalization property places acceptable densities within the intersection of L1 and L3 – a superset of 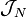 .
.
Topology
The ground state electronic density of an atom is conjectured to be a monotonically decaying function of the distance from the nucleus.[3]
Nuclear cusp condition
The electronic density displays cusps at each nucleus in a molecule as a result of the unbounded electron-nucleus Coulomb potential. This behavior is quantified by the Kato cusp condition formulated in terms of the spherically averaged density, 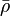 , about any given nucleus as[4]
, about any given nucleus as[4]
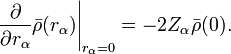
That is, the radial derivative of the spherically averaged density, evaluated at any nucleus, is equal to twice the density at that nucleus multiplied by the negative of the atomic number (Z).
Asymptotic behavior
The nuclear cusp condition provides the near-nuclear (small r) density behavior as
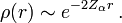
The long-range (large r) behavior of the density is also known, taking the form[5]
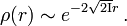
where I is the ionization energy of the system.
Response Density
Another more-general definition of a density is the "linear-response density".[6][7] This is the density that when contracted with any spin-free, one-electron operator yields the associated property defined as the derivative of the energy. For example, a dipole moment is the derivative of the energy with respect to an external magnetic field and is not the expectation value of the operator over the wavefunction. For some theories they are the same when the wavefunction is converged. The occupation numbers are not limited to the range of zero to two, and therefore sometimes even the response density can be negative in certain regions of space.[8]
See also
References
- ↑ Parr, Robert G.; Yang, Weitao (1989). Density-Functional Theory of Atoms and Molecules. New York: Oxford University Press. ISBN 0-19-509276-7.
- ↑ Lieb, Elliott H. (1983). "Density functionals for coulomb systems". International Journal of Quantum Chemistry 24 (3): 243–277. doi:10.1002/qua.560240302.
- ↑ Ayers, Paul W.; Parr, Robert G. (2003). "Sufficient condition for monotonic electron density decay in many-electron systems". International Journal of Quantum Chemistry 95 (6): 877–881. doi:10.1002/qua.10622.
- ↑ Kato, Tosio (1957). "On the eigenfunctions of many-particle systems in quantum mechanics". Communications on Pure and Applied Mathematics 10 (2): 151–177. doi:10.1002/cpa.3160100201.
- ↑ Morrell, Marilyn M.; Parr, Robert. G.; Levy, Mel (1975). "Calculation of ionization potentials from density matrices and natural functions, and the long-range behavior of natural orbitals and electron density". Journal of Chemical Physics 62 (2): 549–554. Bibcode:1975JChPh..62..549M. doi:10.1063/1.430509.
- ↑ Handy, Nicholas C.; Schaefer, Henry F. (1984). "On the evaluation of analytic energy derivatives for correlated wave functions". The Journal of Chemical Physics 81 (11): 5031. Bibcode:1984JChPh..81.5031H. doi:10.1063/1.447489.
- ↑ Wiberg, Kenneth B.; Hadad, Christopher M.; Lepage, Teresa J.; Breneman, Curt M.; Frisch, Michael J. (1992). "Analysis of the effect of electron correlation on charge density distributions". The Journal of Physical Chemistry 96 (2): 671. doi:10.1021/j100181a030.
- ↑ Gordon, Mark S.; Schmidt, Michael W.; Chaban, Galina M.; Glaesemann, Kurt R.; Stevens, Walter J.; Gonzalez, Carlos (1999). "A natural orbital diagnostic for multiconfigurational character in correlated wave functions". J. Chem. Phys. 110 (9): 4199–4207. Bibcode:1999JChPh.110.4199G. doi:10.1063/1.478301.