Drug design
Drug design, sometimes referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target.[1] The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of small molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques.[2] This type of modeling is often referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design.
The phrase "drug design" is to some extent a misnomer. A more accurate term is ligand design (i.e., design of a small molecule that will bind tightly to its target).[3] Although modeling techniques for prediction of binding affinity are reasonably successful, there are many other properties, such as bioavailability, metabolic half-life, side effects, etc., that first must be optimized before a ligand can become a safe and efficacious drug. These other characteristics are often difficult to predict using rational drug design techniques.
Background
Typically a Biomolecular target (proteins or nucleic acids) is a key molecule involved in a particular metabolic or signaling pathway that is leading to a specific disease condition or pathology or to the infectivity or survival of a microbial pathogen. In Some cases, small molecules will be designed to inhibit the target function in the specific pathway (diseased state). Small molecules (inhibitors or modulators) will be designed that are complementary to the active site/allosteric site of target. In some other cases, small molecules will be designed or developed to enhance the normal pathway by promoting specific biomolecular molecules in the normal pathways that may have been affected in the diseased state. Small molecules (drugs) can be designed so as not to affect any other important "off-target" molecules or antitargets, since drug interactions with off-target molecules may lead to undesirable side effects. Sequence homology is often used to identify such risks.
Most commonly, drugs are organic small molecules produced through chemical synthesis, but biopolymer-based drugs (also known as biologics) produced through biological processes are becoming increasingly more common. In addition, mRNA-based gene silencing technologies may have therapeutic applications.
Types

There are two major types of drug design. The first is referred to as ligand-based drug design and the second, structure-based drug design.
Ligand-based
Ligand-based drug design (or indirect drug design) relies on knowledge of other molecules that bind to the biological target of interest. These other molecules may be used to derive a pharmacophore model that defines the minimum necessary structural characteristics a molecule must possess in order to bind to the target.[4] In other words, a model of the biological target may be built based on the knowledge of what binds to it, and this model in turn may be used to design new molecular entities that interact with the target. Alternatively, a quantitative structure-activity relationship (QSAR), in which a correlation between calculated properties of molecules and their experimentally determined biological activity, may be derived. These QSAR relationships in turn may be used to predict the activity of new analogs.
Structure-based
Structure-based drug design (or direct drug design) relies on knowledge of the three dimensional structure of the biological target obtained through methods such as x-ray crystallography or NMR spectroscopy.[5] If an experimental structure of a target is not available, it may be possible to create a homology model of the target based on the experimental structure of a related protein. Using the structure of the biological target, candidate drugs that are predicted to bind with high affinity and selectivity to the target may be designed using interactive graphics and the intuition of a medicinal chemist. Alternatively various automated computational procedures may be used to suggest new drug candidates.
The 3D structures of biomolecular targets are obtained from X-ray crystallography and NMR. In parallel, information about the structural dynamics and electronic properties about ligands are obtained from calculations. This has encouraged the rapid development of the structure-based drug design. Current methods for structure-based drug design can be divided roughly into two categories. The first category is about “finding” ligands for a given receptor, which is usually referred as database searching. In this case, a large number of potential ligand molecules are screened to find those fitting the binding pocket of the receptor. This method is usually referred as ligand-based drug design. The key advantage of database searching is that it saves synthetic effort to obtain new lead compounds. Another category of structure-based drug design methods is about “building” ligands, which is usually referred as receptor-based drug design. In this case, ligand molecules are built up within the constraints of the binding pocket by assembling small pieces in a stepwise manner. These pieces can be either individual atoms or molecular fragments. The key advantage of such a method is that novel structures, not contained in any database, can be suggested.[6][7][8]
Active site identification
Active site identification is the first step in this program. It analyzes the protein to find the binding pocket, derives key interaction sites within the binding pocket, and then prepares the necessary data for Ligand fragment link. The basic inputs for this step are the 3D structure of the protein and a pre-docked ligand in PDB format, as well as their atomic properties. Both ligand and protein atoms need to be classified and their atomic properties should be defined, basically, into four atomic types:
- hydrophobic atom: All carbons in hydrocarbon chains or in aromatic groups.
- H-bond donor: Oxygen and nitrogen atoms bonded to hydrogen atom(s).
- H-bond acceptor: Oxygen and sp2 or sp hybridized nitrogen atoms with lone electron pair(s).
- Polar atom: Oxygen and nitrogen atoms that are neither H-bond donor nor H-bond acceptor, sulfur, phosphorus, halogen, metal, and carbon atoms bonded to hetero-atom(s).
The space inside the ligand binding region would be studied with virtual probe atoms of the four types above so the chemical environment of all spots in the ligand binding region can be known. Hence we are clear what kind of chemical fragments can be put into their corresponding spots in the ligand binding region of the receptor.
Ligand fragment link

A fragments database can enable drug design. The term “fragment” refers to functional groups or portions of molecules which might have bioactivity. Organic molecules can be decomposed into basic chemical fragments. The number of kinds of fragment structures is limited.
There are a large number of possible fragment combinations. A small perturbation of the previous fragment conformation would cause great difference in activity. In order to find the lowest binding energy on the Potential energy surface (PES) between fragments and a receptor pocket, the scoring function calculation would be performed for every step of conformation change of the fragments derived from every type of possible fragments combination. Since this requires a large amount of computation, using different tricks may use less computing power and let the program work more efficiently. When a ligand is inserted into the pocket site of a receptor, groups on the ligand that bind tightly with the receptor should have the highest priority in finding their lowest-energy conformation. This allows us to put several seeds into the program at the same time and optimize the conformation of those seeds that form significant interactions with the receptor, and then connect those seeds into a continuous ligand in a manner that make the rest of the ligand have the lowest energy. The pre-placed seeds ensure high binding affinity and their optimal conformation determines the manner in which the ligand will be built, thus determining the overall structure of the final ligand. This strategy efficiently reduces the calculation burden for fragment construction. On the other hand, it reduces the possibility of the combination of fragments, which reduces the number of possible ligands that can be derived from the program. The two strategies above are widely used in most structure-based drug design programs. They are described as “Grow” and “Link”. The two strategies are always combined in order to make the construction result more reliable.[6][7][9]
Scoring functions
Structure-based drug design attempts to use the structure of proteins as a basis for designing new ligands by applying accepted principles of molecular recognition. The basic assumption underlying structure-based drug design is that a good ligand molecule should bind tightly to its target. Thus, one of the most important principles for designing or obtaining potential new ligands is to predict the binding affinity of a certain ligand to its target and use it as a criterion for selection.
One early method was developed by Böhm[10] to develop a general-purposed empirical scoring function in order to describe the binding energy. The following “Master Equation” was derived:
![\begin{array}{lll}\Delta G_{\text{bind}} = -RT \ln K_{\text{d}}\\[1.3ex]
K_{\text{d}} = \dfrac{[\text{Receptor}][\text{Acceptor}]}{[\text{Complex}]}\\[1.3ex]
\Delta G_{\text{bind}} = \Delta G_{\text{desolvation}} + \Delta G_{\text{motion}} + \Delta G_{\text{configuration}} + \Delta G_{\text{interaction}}\end{array}](../I/m/08eb5c19a07998847fe0aefabd5b3eba.png)
where:
- desolvation – enthalpic penalty for removing the ligand from solvent
- motion – entropic penalty for reducing the degrees of freedom when a ligand binds to its receptor
- configuration – conformational strain energy required to put the ligand in its "active" conformation
- interaction – enthalpic gain for "resolvating" the ligand with its receptor
The basic idea is that the overall binding free energy can be decomposed into independent components that are known to be important for the binding process. Each component reflects a certain kind of free energy alteration during the binding process between a ligand and its target receptor. The Master Equation is the linear combination of these components. According to Gibbs free energy equation, the relation between dissociation equilibrium constant, Kd, and the components of free energy was built.
Various computational methods are used to estimate each of the components of the master equation. For example, the change in polar surface area upon ligand binding can be used to estimate the desolvation energy. The number of rotatable bonds frozen upon ligand binding is proportional to the motion term. The configurational or strain energy can be estimated using molecular mechanics calculations. Finally the interaction energy can be estimated using methods such as the change in non polar surface, statistically derived potentials of mean force, the number of hydrogen bonds formed, etc. In practice, the components of the master equation are fit to experimental data using multiple linear regression. This can be done with a diverse training set including many types of ligands and receptors to produce a less accurate but more general "global" model or a more restricted set of ligands and receptors to produce a more accurate but less general "local" model.[11][12][13]
Rational drug discovery
In contrast to traditional methods of drug discovery, which rely on trial-and-error testing of chemical substances on cultured cells or animals, and matching the apparent effects to treatments, rational drug design begins with a hypothesis that modulation of a specific biological target may have therapeutic value. In order for a biomolecule to be selected as a drug target, two essential pieces of information are required. The first is evidence that modulation of the target will have therapeutic value. This knowledge may come from, for example, disease linkage studies that show an association between mutations in the biological target and certain disease states. The second is that the target is "drugable". This means that it is capable of binding to a small molecule and that its activity can be modulated by the small molecule.
Once a suitable target has been identified, the target is normally cloned and expressed. The expressed target is then used to establish a screening assay. In addition, the three-dimensional structure of the target may be determined.
The search for small molecules that bind to the target is begun by screening libraries of potential drug compounds. This may be done by using the screening assay (a "wet screen"). In addition, if the structure of the target is available, a virtual screen may be performed of candidate drugs. Ideally the candidate drug compounds should be "drug-like", that is they should possess properties that are predicted to lead to oral bioavailability, adequate chemical and metabolic stability, and minimal toxic effects. Several methods are available to estimate druglikeness such as Lipinski's Rule of Five and a range of scoring methods such as Lipophilic efficiency. Several methods for predicting drug metabolism have been proposed in the scientific literature, and a recent example is SPORCalc.[14] Due to the complexity of the drug design process, two terms of interest are still serendipity and bounded rationality. Those challenges are related to the large amount of chemical space that any potential new drugs must have if they are not to have unacceptable adverse effects.
Applications of Computer-aided drug design
The most fundamental goal is to predict whether a given molecule will bind to a target and if so how strongly. Molecular mechanics or molecular dynamics are most often used to predict the conformation of the small molecule and to model conformational changes in the biological target that may occur when the small molecule binds to it. Semi-empirical, ab initio quantum chemistry methods, or density functional theory are often used to provide optimized parameters for the molecular mechanics calculations and also provide an estimate of the electronic properties (electrostatic potential, polarizability, etc.) of the drug candidate that will influence binding affinity.
Molecular mechanics methods may also be used to provide semi-quantitative prediction of the binding affinity. Also, knowledge-based scoring function may be used to provide binding affinity estimates. These methods use linear regression, machine learning, neural nets or other statistical techniques to derive predictive binding affinity equations by fitting experimental affinities to computationally derived interaction energies between the small molecule and the target.[15][16]
Ideally, the computational method will be able to predict affinity before a compound is synthesized and hence in theory only one compound needs to be synthesized, saving enormous time and cost. The reality is that present computational methods are imperfect and provide, at best, only qualitatively accurate estimates of affinity. In practice it still takes several iterations of design, synthesis, and testing before an optimal drug is discovered. Computational methods have accelerated discovery by reducing the number of iterations required and have often provided novel structures.[17][18]
Drug design with the help of computers may be used at any of the following stages of drug discovery:
- hit identification using virtual screening (structure- or ligand-based design)
- hit-to-lead optimization of affinity and selectivity (structure-based design, QSAR, etc.)
- lead optimization optimization of other pharmaceutical properties while maintaining affinity
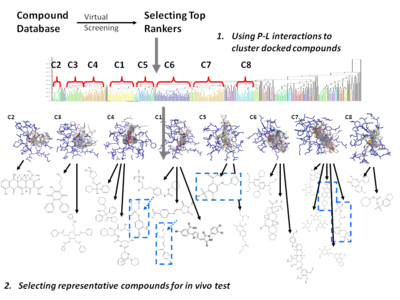
In order to overcome the insufficient prediction of binding affinity calculated by recent scoring functions, the protein-ligand interaction and compound 3D structure information are used for analysis. For structure-based drug design, several post-screening analyses focusing on protein-ligand interaction have been developed for improving enrichment and effectively mining potential candidates:
- Consensus scoring[19][20]
- Selecting candidates by voting of multiple scoring functions
- May lose the relationship between protein-ligand structural information and scoring criterion
- Geometric analysis
- Comparing protein-ligand interactions by visually inspecting individual structures
- Becoming intractable when the number of complexes to be analyzed increasing
- Cluster analysis[21][22]
- Represent and cluster candidates according to protein-ligand 3D information
- Needs meaningful representation of protein-ligand interactions.
Examples
A particular example of rational drug design involves the use of three-dimensional information about biomolecules obtained from such techniques as X-ray crystallography and NMR spectroscopy. Computer-aided drug design in particular becomes much more tractable when there is a high-resolution structure of a target protein bound to a potent ligand. This approach to drug discovery is sometimes referred to as structure-based drug design. The first unequivocal example of the application of structure-based drug design leading to an approved drug is the carbonic anhydrase inhibitor dorzolamide, which was approved in 1995.[23][24]
Another important case study in rational drug design is imatinib, a tyrosine kinase inhibitor designed specifically for the bcr-abl fusion protein that is characteristic for Philadelphia chromosome-positive leukemias (chronic myelogenous leukemia and occasionally acute lymphocytic leukemia). Imatinib is substantially different from previous drugs for cancer, as most agents of chemotherapy simply target rapidly dividing cells, not differentiating between cancer cells and other tissues.
Additional examples include:
- Many of the atypical antipsychotics
- Cimetidine, the prototypical H2-receptor antagonist from which the later members of the class were developed
- Selective COX-2 inhibitor NSAIDs
- Dorzolamide, a carbonic anhydrase inhibitor used to treat glaucoma
- Enfuvirtide, a peptide HIV entry inhibitor
- Nonbenzodiazepines like zolpidem and zopiclone
- Probenecid
- SSRIs (selective serotonin reuptake inhibitors), a class of antidepressants
- Zanamivir, an antiviral drug
- Isentress, HIV Integrase inhibitor[25]
Case Studies
- 5-HT3 antagonists
- Acetylcholine receptor agonists
- Angiotensin receptor blockers
- Bcr-Abl tyrosine kinase inhibitors
- Cannabinoid receptor antagonists
- CCR5 receptor antagonists
- Cyclooxygenase 2 inhibitors
- Dipeptidyl peptidase-4 inhibitors
- HIV protease inhibitors
- NK1 receptor antagonists
- Non-nucleoside reverse transcriptase inhibitors
- Proton pump inibitors
- Triptans
- TRPV1 antagonists
- Renin inhibitors
- c-Met inhibitors
- PDE5 inhibitors
Criticism
It has been argued that the highly rigid and focused nature of rational drug design suppresses serendipity in drug discovery.[26] Because many of the most significant medical discoveries have been inadvertent, the recent focus on rational drug design may limit the progress of drug discovery. Furthermore, the rational design of a drug may be limited by a crude or incomplete understanding of the underlying molecular processes of the disease it is intended to treat.
See also
References
- ↑ Madsen, Ulf; Krogsgaard-Larsen, Povl; Liljefors, Tommy (2002). Textbook of Drug Design and Discovery. Washington, DC: Taylor & Francis. ISBN 0-415-28288-8.
- ↑ Cohen, N. Claude (1996). Guidebook on Molecular Modeling in Drug Design. Boston: Academic Press. ISBN 0-12-178245-X.
- ↑ Tollenaere JP (April 1996). "The role of structure-based ligand design and molecular modelling in drug discovery". Pharm World Sci 18 (2): 56–62. doi:10.1007/BF00579706. PMID 8739258.
- ↑ Guner, Osman F. (2000). Pharmacophore Perception, Development, and use in Drug Design. La Jolla, Calif: International University Line. ISBN 0-9636817-6-1.
- ↑ Leach, Andrew R.; Harren Jhoti (2007). Structure-based Drug Discovery. Berlin: Springer. ISBN 1-4020-4406-2.
- ↑ 6.0 6.1 Wang R,Gao Y,Lai L (2000). "LigBuilder: A Multi-Purpose Program for Structure-Based Drug Design". Journal of Molecular Modeling 6 (7–8): 498–516. doi:10.1007/s0089400060498.
- ↑ 7.0 7.1 Schneider G, Fechner U (August 2005). "Computer-based de novo design of drug-like molecules". Nat Rev Drug Discov 4 (8): 649–63. doi:10.1038/nrd1799. PMID 16056391.
- ↑ Jorgensen WL (March 2004). "The many roles of computation in drug discovery". Science 303 (5665): 1813–8. Bibcode:2004Sci...303.1813J. doi:10.1126/science.1096361. PMID 15031495.
- ↑ Verlinde CL, Hol WG (July 1994). "Structure-based drug design: progress, results and challenges". Structure 2 (7): 577–87. doi:10.1016/S0969-2126(00)00060-5. PMID 7922037.
- ↑ Böhm HJ (June 1994). "The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure". J. Comput. Aided Mol. Des. 8 (3): 243–56. Bibcode:1994JCAMD...8..243B. doi:10.1007/BF00126743. PMID 7964925.
- ↑ Gohlke H, Hendlich M, Klebe G (January 2000). "Knowledge-based scoring function to predict protein-ligand interactions". J. Mol. Biol. 295 (2): 337–56. doi:10.1006/jmbi.1999.3371. PMID 10623530.
- ↑ Clark RD, Strizhev A, Leonard JM, Blake JF, Matthew JB (January 2002). "Consensus scoring for ligand/protein interactions". J. Mol. Graph. Model. 20 (4): 281–95. doi:10.1016/S1093-3263(01)00125-5. PMID 11858637.
- ↑ Wang R, Lai L, Wang S (January 2002). "Further development and validation of empirical scoring functions for structure-based binding affinity prediction". J. Comput. Aided Mol. Des. 16 (1): 11–26. Bibcode:2002JCAMD..16...11W. doi:10.1023/A:1016357811882. PMID 12197663.
- ↑ Smith J, Stein V (April 2009). "SPORCalc: A development of a database analysis that provides putative metabolic enzyme reactions for ligand-based drug design". Computational Biology and Chemistry 33 (2): 149–59. doi:10.1016/j.compbiolchem.2008.11.002. PMID 19157988.
- ↑ Rajamani R, Good AC (May 2007). "Ranking poses in structure-based lead discovery and optimization: current trends in scoring function development". Curr Opin Drug Discov Devel 10 (3): 308–15. PMID 17554857.
- ↑ de Azevedo WF, Dias R (December 2008). "Computational methods for calculation of ligand-binding affinity". Curr Drug Targets 9 (12): 1031–9. doi:10.2174/138945008786949405. PMID 19128212.
- ↑ Singh J, Chuaqui CE, Boriack-Sjodin PA et al. (December 2003). "Successful shape-based virtual screening: the discovery of a potent inhibitor of the type I TGFbeta receptor kinase (TbetaRI)". Bioorg. Med. Chem. Lett. 13 (24): 4355–9. doi:10.1016/j.bmcl.2003.09.028. PMID 14643325.
- ↑ Becker OM, Dhanoa DS, Marantz Y et al. (June 2006). "An integrated in silico 3D model-driven discovery of a novel, potent, and selective amidosulfonamide 5-HT1A agonist (PRX-00023) for the treatment of anxiety and depression". J. Med. Chem. 49 (11): 3116–35. doi:10.1021/jm0508641. PMID 16722631.
- ↑ Liang S, Meroueh SO, Wang G, Qiu C, Zhou Y (May 2009). "Consensus scoring for enriching near-native structures from protein-protein docking decoys". Proteins 75 (2): 397–403. doi:10.1002/prot.22252. PMC 2656599. PMID 18831053.
- ↑ Oda A, Tsuchida K, Takakura T, Yamaotsu N, Hirono S (2006). "Comparison of consensus scoring strategies for evaluating computational models of protein-ligand complexes". J Chem Inf Model 46 (1): 380–91. doi:10.1021/ci050283k. PMID 16426072.
- ↑ Deng Z, Chuaqui C, Singh J (January 2004). "Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions". J. Med. Chem. 47 (2): 337–44. doi:10.1021/jm030331x. PMID 14711306.
- ↑ Amari S, Aizawa M, Zhang J, Fukuzawa K, Mochizuki Y, Iwasawa Y, Nakata K, Chuman H, Nakano T (2006). "VISCANA: visualized cluster analysis of protein-ligand interaction based on the ab initio fragment molecular orbital method for virtual ligand screening". J Chem Inf Model 46 (1): 221–30. doi:10.1021/ci050262q. PMID 16426058.
- ↑ Greer J, Erickson JW, Baldwin JJ, Varney MD (April 1994). "Application of the three-dimensional structures of protein target molecules in structure-based drug design". Journal of Medicinal Chemistry 37 (8): 1035–54. doi:10.1021/jm00034a001. PMID 8164249.
- ↑ Hendrik Timmerman; Klaus Gubernator; Hans-Joachim Böhm; Raimund Mannhold; Hugo Kubinyi (1998). Structure-based Ligand Design (Methods and Principles in Medicinal Chemistry). Weinheim: Wiley-VCH. ISBN 3-527-29343-4.
- ↑ "AutoDock's role in Developing the First Clinically-Approved HIV Integrase Inhibitor". Press Release. The Scripps Research Institute. 2007-12-17.
- ↑ Klein DF (2008). "The loss of serendipity in psychopharmacology". JAMA 299 (9): 1063–5. doi:10.1001/jama.299.9.1063. PMID 18319418.
External links
- Drug Design at the US National Library of Medicine Medical Subject Headings (MeSH)
| ||||||||||
| ||||||