Didanosine
 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
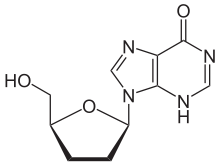
| 9-((2R,5S)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-3H-purin-6(9H)-one | |
| Clinical data | |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a691006 |
| |
| Oral | |
| Pharmacokinetic data | |
| Bioavailability | 30 to 54% |
| Protein binding | Less than 5% |
| Half-life | 1.5 hours |
| Excretion | Renal |
| Identifiers | |
|
69655-05-6 | |
| J05AF02 | |
| PubChem | CID 50599 |
| DrugBank |
DB00900 |
| ChemSpider |
45864 |
| UNII |
K3GDH6OH08 |
| KEGG |
D00296 |
| ChEBI |
CHEBI:490877 |
| ChEMBL |
CHEMBL1460 |
| NIAID ChemDB | 000004 |
| Chemical data | |
| Formula | C10H12N4O3 |
| 236.227 g/mol | |
|
SMILES
| |
| |
| | |
Didanosine (2′,3′-dideoxyinosine, ddI, DDI) marketed under the trade names Videx and Videx EC. It is an antiretroviral medication used to treat HIV/AIDS in combination with other medications as part of highly active antiretroviral therapy (HAART). It is of the reverse transcriptase inhibitor class.
It is on the World Health Organization's List of Essential Medicines, a list of the most important medication needed in a basic health system.[1]
Adverse effects
The most common adverse events with didanosine are diarrhea, nausea, vomiting, abdominal pain, fever, headache and rash. Peripheral neuropathy occurred in 21-26% of participants in key didanosine trials.[2]
Pancreatitis is rarely observed but has caused occasional fatalities, and has black box warning status. Other reported serious adverse events are retinal changes, optic neuritis and alterations of liver functions. The risk of some of these serious adverse events is increased by drinking alcohol.
In February 2010, the United States Food and Drug Administration issued a statement that patients using Didanosine (Videx) are at risk for a rare but potentially fatal liver disorder, non-cirrhotic portal hypertension.[3]
Drug interactions
- A significant interaction has also been recorded with allopurinol, and administration of these drugs together should be avoided.[2]
- Indinavir and delavirdine show reduced in plasma levels when administered simultaneously with didanosine; these drugs should be administered at different times.[2]
- Ketoconazole, itraconazole, ciprofloxacin should be administered at a different time from didanosine due to interactions with the buffering agent.[2]
- Administration with drugs with overlapping toxicity, such as zalcitabine and stavudine, is not recommended.[4]
- Alcohol can exacerbate didanosine's toxicity, and avoiding drinking alcohol while taking didanosine is recommended.[2]
Resistance
Drug resistance to didanosine does develop, though slower than to zidovudine (AZT). The most common mutation observed in vivo is L74V in the viral pol gene, which confers cross-resistance to zalcitabine; other mutations observed include K65R and M184V .[2][5]
Mechanism of action
Didanosine (ddI) is a nucleoside analogue of guanosine. It differs from other nucleoside analogues, because it does not have any of the regular bases, instead it has hypoxanthine attached to the sugar ring. Within the cell, ddI is phosphorylated to the active metabolite of dideoxyadenosine triphosphate, ddATP, by cellular enzymes. Like other anti-HIV nucleoside analogs, it acts as a chain terminator by incorporation and inhibits viral reverse transcriptase by competing with natural dATP.
Pharmacokinetics
Oral absorption of didanosine is fairly low (42%)[2] but rapid. Food substantially reduces didanosine bioavailability, and the drug should be administered on an empty stomach.[2] The half-life in plasma is only 1.5 hours,[2] but in the intracellular environment more than 12 hours. An enteric-coated formulation is now marketed as well. Elimination is predominantly renal; the kidneys actively secrete didanosine, the amount being 20% of the oral dose.
History
The related pro-drug of didanosine, 2′,3′-dideoxyadenosine (ddA), was initially synthesized by Morris J. Robins (professor of Organic Chemistry at Brigham Young University) and R.K. Robins in 1964. Subsequently, Samuel Broder, Hiroaki Mitsuya, and Robert Yarchoan in the National Cancer Institute (NCI) found that ddA and ddI could inhibit HIV replication in the test tube and conducted initial clinical trials showing that didanosine had activity in patients infected with HIV. On behalf of the NCI, they were awarded patents on these activities. Since the NCI does not market products directly, the National Institutes of Health (NIH) awarded a ten-year exclusive license to Bristol-Myers Squibb Co. (BMS) to market and sell ddI as Videx tablets.
Didanosine became the second drug approved for the treatment of HIV infection in many other countries, including in the United States by the Food and Drug Administration (FDA) on October 9, 1991. Its FDA approval helped bring down the price of zidovudine (AZT), the initial anti-HIV drug.
Didanosine has weak acid stability and is easily damaged by stomach acid. Therefore, the original formula approved by the FDA used chewable tablets that included an antacid buffering compound to neutralize stomach acid. The chewable tablets were not only large and fragile, they also were foul-tasting and the buffering compound would cause diarrhea. Although the FDA had not approved the original formulation for once-a-day dosing it was possible for some people to take it that way.
At the end of its ten-year license, BMS re-formulated Videx as Videx EC and patented that, which reformulation the FDA approved in 2000. The new formulation is a smaller capsule containing coated microspheres instead of using a buffering compound. It is approved by the FDA for once-a-day dosing. Also at the end of that ten-year period, the NIH licensed didanosine to Barr Laboratories under a non-exclusive license, and didanosine became the first generic anti-HIV drug marketed in the United States.
One of the patents for ddI expired in the United States on August 29, 2006, but other patents extend beyond that time.
Sources
- ↑ "WHO Model List of EssentialMedicines". World Health Organization. October 2013. Retrieved 22 April 2014.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 VIDEX (didanosine): chewable/dispersible buffered tablets; buffered powder for oral solution; pediatric powder for oral solution. Product information (July 2000)
- ↑ http://hosted.ap.org/dynamic/stories/U/US_HIV_DRUG_LIVER_RISKS?SITE=KYB66&SECTION=HOME&TEMPLATE=DEFAULT
- ↑ DHHS panel. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents (May 4, 2006). (Available for download from AIDSInfo)
- ↑ Moyle GJ. Use of viral resistance patterns to antiretroviral drugs in optimising selection of drug combinations and sequences. Drugs 1996;52:168-185
Further reading
- Robins MJ, McCarthy JR Jr., Robins RJ. Biochemistry 1966; 5(1):224-31.
- Yarchoan R, Mitsuya H, Broder S. AIDS therapies. Sci Am 1988;259(4):110-9.
- Männistö P.T., Tuominen R.K. in Farmakologia ja Toksikologia, 5th edition: (ed. Koulu, Tuomisto, Paasonen) Medicina, 1996.
- Rang H.P., Dale M.M., Ritter J.M.: Pharmacology, 3rd edition. Pearson Professional Ltd, 1995.
- Watson et al.: Molecular Biology of the Gene 4th edition. The Benjamin/Cummings Publishing Company, 1987.
- Mitsuya H, Yarchoan R, Broder S. Molecular targets for AIDS therapy. Science 1990;249(4976):1533-44.
- Yarchoan R, Mitsuya H, Thomas RV, et al. In vivo activity against HIV and favorable toxicity profile of 2',3'-dideoxyinosine. Science 1989;245(4916):412-5.
- NIH Oral History of Samuel Broder describing development of AIDS drugs
- NIH Oral History of Robert Yarchoan describing development of AIDS drugs
- NIH Office of Technology Transfer Report on Development and Licensing of ddI
- FDA reviews safety of two HIV drugs