Detailed balance
The principle of detailed balance is formulated for kinetic systems which are decomposed into elementary processes (collisions, or steps, or elementary reactions): At equilibrium, each elementary process should be equilibrated by its reverse process.
History
The principle of detailed balance was explicitly introduced for collisions by Ludwig Boltzmann. In 1872, he proved his H-theorem using this principle.[1] The arguments in favor of this property are founded upon microscopic reversibility.[2]
Five years before Boltzmann, James Clerk Maxwell used the principle of detailed balance for gas kinetics with the reference to the principle of sufficient reason.[3] He compared the idea of detailed balance with other types of balancing (like cyclic balance) and found that "Now it is impossible to assign a reason" why detailed balance should be rejected (pg. 64).
Albert Einstein in 1916 used the principle of detailed balance in a background for his quantum theory of emission and absorption of radiation.[4]
In 1901, Rudolf Wegscheider introduced the principle of detailed balance for chemical kinetics.[5] In particular, he demonstrated that the irreversible cycles 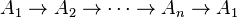 are impossible and found explicitly the relations between kinetic constants that follow from the principle of detailed balance. In 1931, Lars Onsager used these relations in his works,[6] for which he was awarded the 1968 Nobel Prize in Chemistry.
are impossible and found explicitly the relations between kinetic constants that follow from the principle of detailed balance. In 1931, Lars Onsager used these relations in his works,[6] for which he was awarded the 1968 Nobel Prize in Chemistry.
The principle of detailed balance has been used in Markov chain Monte Carlo methods since their invention in 1953.[7] In particular, in the Metropolis–Hastings algorithm and in its important particular case, Gibbs sampling, it is used as a simple and reliable condition to provide the desirable equilibrium state.
Now, the principle of detailed balance is a standard part of the university courses in statistical mechanics, physical chemistry, chemical and physical kinetics.[8][9][10]
Microscopical background
The microscopic "reversing of time" turns at the kinetic level into the "reversing of arrows": the elementary processes transform into their reverse processes. For example, the reaction
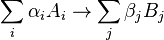 transforms into
transforms into 
and conversely. (Here, 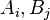 are symbols of components or states,
are symbols of components or states, 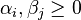 are coefficients). The equilibrium ensemble should be invariant with respect to this transformation because of microreversibility and the uniqueness of thermodynamic equilibrium. This leads us immediately to the concept of detailed balance: each process is equilibrated by its reverse process.
are coefficients). The equilibrium ensemble should be invariant with respect to this transformation because of microreversibility and the uniqueness of thermodynamic equilibrium. This leads us immediately to the concept of detailed balance: each process is equilibrated by its reverse process.
This reasoning is based on three assumptions:
-
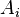 does not change under time reversal;
does not change under time reversal; - Equilibrium is invariant under time reversal;
- The macroscopic elementary processes are microscopically distinguishable. That is, they represent disjoint sets of microscopic events.
Any of these assumptions may be violated.[11] For example, Boltzmann's collision can be represented as 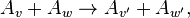 where
where 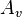 is a particle with velocity v. Under time reversal transforms into
is a particle with velocity v. Under time reversal transforms into 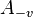 . Therefore, the collision is transformed into the reverse collision by the PT transformation, where P is the space inversion and T is the time reversal. Detailed balance for Boltzmann's equation requires PT-invariance of collisions' dynamics, not just T-invariance. The laws of mechanics are both T- and P-invariant.
. Therefore, the collision is transformed into the reverse collision by the PT transformation, where P is the space inversion and T is the time reversal. Detailed balance for Boltzmann's equation requires PT-invariance of collisions' dynamics, not just T-invariance. The laws of mechanics are both T- and P-invariant.
Equilibrium may be not T- or PT-invariant even if the laws of motion are invariant. This non-invariance may be caused by the spontaneous symmetry breaking. There exist nonreciprocal media (for example, some bi-isotropic materials) without T and PT invariance.[11]
If different macroscopic processes are sampled from the same elementary microscopic events then macroscopic detailed balance may be violated even when microscopic detailed balance holds.[11][12]
Now, after almost 150 years of development, the scope of validity and the violations of detailed balance in kinetics seem to be clear.
Reversible Markov chains
A Markov process is called a reversible Markov process or reversible Markov chain precisely if it satisfies the detailed balance equations.[13] These equations require that the transition probability matrix, P, for the Markov process possess a stationary distribution ((i.e. equilibrium probability distribution) π such that
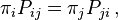
where Pij is the Markov transition probability) from state i to state j, i.e. Pij = P(Xt = j | Xt − 1 = i), and πi and πj are the equilibrium probabilities of being in states i and j, respectively.[13] When Pr(Xt−1 = i) = πi for all i, this is equivalent to the joint probability matrix, Pr(Xt−1 = i, Xt = j) being symmetric in i and j; or symmetric in t − 1 and t.
The definition carries over straightforwardly to continuous variables, where π becomes a probability density, and P(s′, s) a transition kernel probability density from state s′ to state s:
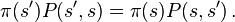
The detailed balance condition is stronger than that required merely for a stationary distribution; that is, there are Markov processes with stationary distributions that do not have detailed balance. Detailed balance implies that, around any closed cycle of states, there is no net flow of probability. For example, it implies that, for all a, b and c,
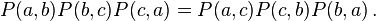
This can be proved by substitution from the definition. In the case of a positive transition matrix, the "no net flow" condition implies detailed balance. Indeed, a necessary and sufficient condition for the reversibility condition is Kolmogorov's criterion, which demands that for the reversible chains the product of transition rates over any closed loop of states must be the same in both directions.
Transition matrices that are symmetric (Pij = Pji or P(s′, s) = P(s, s′)) always have detailed balance. In these cases, a uniform distribution over the states is an equilibrium distribution. For continuous systems with detailed balance, it may be possible to continuously transform the coordinates until the equilibrium distribution is uniform, with a transition kernel which then is symmetric. In the case of discrete states, it may be possible to achieve something similar by breaking the Markov states into appropriately-sized degenerate sub-states.
Detailed balance and entropy increase
For many systems of physical and chemical kinetics, detailed balance provides sufficient conditions for the strict increase of entropy in isolated systems. For example, the famous Boltzmann H-theorem[1] states that, according to the Boltzmann equation, the principle of detailed balance implies positivity of entropy production. The Boltzmann formula (1872) for entropy production in rarefied gas kinetics with detailed balance[1][2] served as a prototype of many similar formulas for dissipation in mass action kinetics[14] and generalized mass action kinetics[15] with detailed balance.
Nevertheless, the principle of detailed balance is not necessary for entropy growth. For example, in the linear irreversible cycle 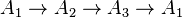 , entropy production is positive but the principle of detailed balance does not hold.
, entropy production is positive but the principle of detailed balance does not hold.
Thus, the principle of detailed balance is a sufficient but not necessary condition for entropy increase in Boltzmann kinetics. These relations between the principle of detailed balance and the second law of thermodynamics were clarified in 1887 when Hendrik Lorentz objected to the Boltzmann H-theorem for polyatomic gases.[16] Lorentz stated that the principle of detailed balance is not applicable to collisions of polyatomic molecules.
Boltzmann immediately invented a new, more general condition sufficient for entropy growth.[17] Boltzmann's condition holds for all Markov processes, irrespective of time-reversibility. Later, entropy increase was proved for all Markov processes by a direct method.[18][19] These theorems may be considered as simplifications of the Boltzmann result. Later, this condition was referred to as the "cyclic balance" condition (because it holds for irreversible cycles) or the "semi-detailed balance" or the "complex balance". In 1981, Carlo Cercignani and Maria Lampis proved that the Lorentz arguments were wrong and the principle of detailed balance is valid for polyatomic molecules.[20] Nevertheless, the extended semi-detailed balance conditions invented by Boltzmann in this discussion remain the remarkable generalization of the detailed balance.
Wegscheider's conditions for the generalized mass action law
In chemical kinetics, the elementary reactions are represented by the stoichiometric equations
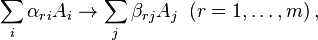
where are the components and  are the stoichiometric coefficients. Here, the reverse reactions with positive constants are included in the list separately. We need this separation of direct and reverse reactions to apply later the general formalism to the systems with some irreversible reactions. The system of stoichiometric equations of elementary reactions is the reaction mechanism.
are the stoichiometric coefficients. Here, the reverse reactions with positive constants are included in the list separately. We need this separation of direct and reverse reactions to apply later the general formalism to the systems with some irreversible reactions. The system of stoichiometric equations of elementary reactions is the reaction mechanism.
The stoichiometric matrix is  ,
, 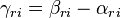 (gain minus loss). The stoichiometric vector
(gain minus loss). The stoichiometric vector 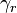 is the rth row of
is the rth row of 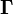 with coordinates .
with coordinates .
According to the generalized mass action law, the reaction rate for an elementary reaction is
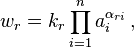
where 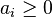 is the activity (the "effective concentration") of .
is the activity (the "effective concentration") of .
The reaction mechanism includes reactions with the reaction rate constants  . For each r the following notations are used:
. For each r the following notations are used:  ;
; 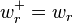 ;
; 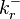 is the reaction rate constant for the reverse reaction if it is in the reaction mechanism and 0 if it is not;
is the reaction rate constant for the reverse reaction if it is in the reaction mechanism and 0 if it is not;  is the reaction rate for the reverse reaction if it is in the reaction mechanism and 0 if it is not. For a reversible reaction,
is the reaction rate for the reverse reaction if it is in the reaction mechanism and 0 if it is not. For a reversible reaction,  is the equilibrium constant.
is the equilibrium constant.
The principle of detailed balance for the generalized mass action law is: For given values 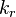 there exists a positive equilibrium
there exists a positive equilibrium 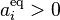 that satisfies detailed balance, that is,
that satisfies detailed balance, that is, 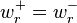 . This means that the system of linear detailed balance equations
. This means that the system of linear detailed balance equations
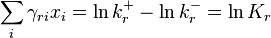
is solvable (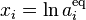 ). The following classical result gives the necessary and sufficient conditions for the existence of a positive equilibrium with detailed balance (see, for example, the textbook[9]).
). The following classical result gives the necessary and sufficient conditions for the existence of a positive equilibrium with detailed balance (see, for example, the textbook[9]).
Two conditions are sufficient and necessary for solvability of the system of detailed balance equations:
- If
 then
then 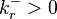 and, conversely, if then (reversibility);
and, conversely, if then (reversibility); - For any solution
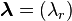 of the system
of the system
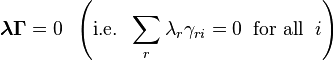
the Wegscheider's identity[21] holds:

Remark. It is sufficient to use in the Wegscheider conditions a basis of solutions of the system 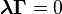 .
.
In particular, for any cycle in the monomolecular (linear) reactions the product of the reaction rate constants in the clockwise direction is equal to the product of the reaction rate constants in the counterclockwise direction. The same condition is valid for the reversible Markov processes (it is equivalent to the "no net flow" condition).
A simple nonlinear example gives us a linear cycle supplemented by one nonlinear step:[21]

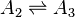
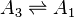

There are two nontrivial independent Wegscheider's identities for this system:
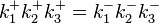 and
and 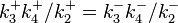
They correspond to the following linear relations between the stoichiometric vectors:
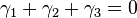 and
and 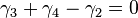 .
.
The computational aspect of the Wegscheider conditions was studied by D. Colquhoun with co-authors.[22]
The Wegscheider conditions demonstrate that whereas the principle of detailed balance states a local property of equilibrium, it implies the relations between the kinetic constants that are valid for all states far from equilibrium. This is possible because a kinetic law is known and relations between the rates of the elementary processes at equilibrium can be transformed into relations between kinetic constants which are used globally. For the Wegscheider conditions this kinetic law is the law of mass action (or the generalized law of mass action).
Dissipation in systems with detailed balance
To describe dynamics of the systems that obey the generalized mass action law, one has to represent the activities as functions of the concentrations cj and temperature. For this purpose, use the representation of the activity through the chemical potential:

where μi is the chemical potential of the species under the conditions of interest, μoi is the chemical potential of that species in the chosen standard state, R is the gas constant and T is the thermodynamic temperature.
The chemical potential can be represented as a function of c and T, where c is the vector of concentrations with components cj. For the ideal systems, 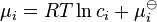 and
and 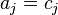 : the activity is the concentration and the generalized mass action law is the usual law of mass action.
: the activity is the concentration and the generalized mass action law is the usual law of mass action.
Let us consider a system in isothermal (T=const) isochoric (the volume V=const) condition. For these conditions, the Helmholtz free energy F(T,V,N) measures the “useful” work obtainable from a system. It is a functions of the temperature T, the volume V and the amounts of chemical components Nj (usually measured in moles), N is the vector with components Nj. For the ideal systems, 
The chemical potential is a partial derivative:  .
.
The chemical kinetic equations are
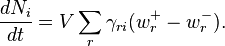
If the principle of detailed balance is valid then for any value of T there exists a positive point of detailed balance ceq:
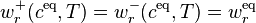
Elementary algebra gives

where 
For the dissipation we obtain from these formulas:
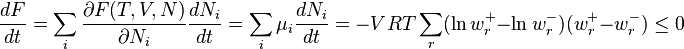
The inequality holds because ln is a monotone function and, hence, the expressions 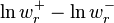 and
and  have always the same sign.
have always the same sign.
Similar inequalities[9] are valid for other classical conditions for the closed systems and the corresponding characteristic functions: for isothermal isobaric conditions the Gibbs free energy decreases, for the isochoric systems with the constant internal energy (isolated systems) the entropy increases as well as for isobaric systems with the constant enthalpy.
Onsager reciprocal relations and detailed balance
Let the principle of detailed balance be valid. Then, for small deviations from equilibrium, the kinetic response of the system can be approximated as linearly related to the its deviation from chemical equilibrium, giving the reaction rates for the generalized mass action law as:
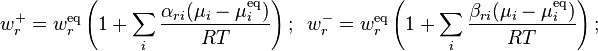
Therefore, again in the linear response regime near equilibrium, the kinetic equations are ():
![\frac{d N_i}{d t}=-V \sum_j \left[\sum_r w^{\rm eq}_r \gamma_{ri}\gamma_{rj}\right] \frac{\mu_j-\mu^{\rm eq}_j}{RT}.](../I/m/d71dc37e0926f149b785981234b7cb45.png)
This is exactly the Onsager form: following the original work of Onsager,[6] we should introduce the thermodynamic forces 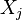 and the matrix of coefficients
and the matrix of coefficients 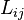 in the form
in the form
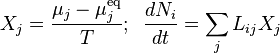
The coefficient matrix is symmetric:

These symmetry relations, 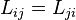 , are exactly the Onsager reciprocal relations. The coefficient matrix
, are exactly the Onsager reciprocal relations. The coefficient matrix 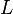 is non-positive. It is negative on the linear span of the stoichiometric vectors
is non-positive. It is negative on the linear span of the stoichiometric vectors 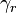 .
.
So, the Onsager relations follow from the principle of detailed balance in the linear approximation near equilibrium.
Semi-detailed balance
To formulate the principle of semi-detailed balance, it is convenient to count the direct and inverse elementary reactions separately. In this case, the kinetic equations have the form:

Let us use the notations 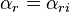 ,
, 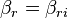 for the input and the output vectors of the stoichiometric coefficients of the rth elementary reaction. Let
for the input and the output vectors of the stoichiometric coefficients of the rth elementary reaction. Let 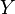 be the set of all these vectors
be the set of all these vectors 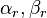 .
.
For each 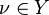 , let us define two sets of numbers:
, let us define two sets of numbers:
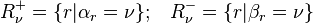
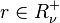 if and only if
if and only if 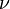 is the vector of the input stoichiometric coefficients
is the vector of the input stoichiometric coefficients 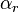 for the rth elementary reaction;
for the rth elementary reaction;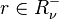 if and only if is the vector of the output stoichiometric coefficients
if and only if is the vector of the output stoichiometric coefficients 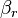 for the rth elementary reaction.
for the rth elementary reaction.
The principle of semi-detailed balance means that in equilibrium the semi-detailed balance condition holds: for every
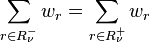
The semi-detailed balance condition is sufficient for the stationarity: it implies that
 .
.
For the Markov kinetics the semi-detailed balance condition is just the elementary balance equation and holds for any steady state. For the nonlinear mass action law it is, in general, sufficient but not necessary condition for stationarity.
The semi-detailed balance condition is weaker than the detailed balance one: if the principle of detailed balance holds then the condition of semi-detailed balance also holds.
For systems that obey the generalized mass action law the semi-detailed balance condition is sufficient for the dissipation inequality 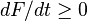 (for the Helmholtz free energy under isothermal isochoric conditions and for the dissipation inequalities under other classical conditions for the corresponding thermodynamic potentials).
(for the Helmholtz free energy under isothermal isochoric conditions and for the dissipation inequalities under other classical conditions for the corresponding thermodynamic potentials).
Boltzmann introduced the semi-detailed balance condition for collisions in 1887[17] and proved that it guaranties the positivity of the entropy production. For chemical kinetics, this condition (as the complex balance condition) was introduced by Horn and Jackson in 1972.[23]
The microscopic backgrounds for the semi-detailed balance were found in the Markov microkinetics of the intermediate compounds that are present in small amounts and whose concentrations are in quasiequilibrium with the main components.[24] Under these microscopic assumptions, the semi-detailed balance condition is just the balance equation for the Markov microkinetics according to the Michaelis–Menten–Stueckelberg theorem.[25]
Dissipation in systems with semi-detailed balance
Let us represent the generalized mass action law in the equivalent form: the rate of the elementary process

is
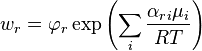
where  is the chemical potential and
is the chemical potential and  is the Helmholtz free energy. The exponential term is called the Boltzmann factor and the multiplier
is the Helmholtz free energy. The exponential term is called the Boltzmann factor and the multiplier 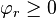 is the kinetic factor.[25]
Let us count the direct and reverse reaction in the kinetic equation separately:
is the kinetic factor.[25]
Let us count the direct and reverse reaction in the kinetic equation separately:
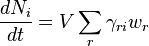
An auxiliary function 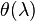 of one variable
of one variable ![\lambda\in [0,1]](../I/m/ada29722c713d4d8a571d9a4ce353ae6.png) is convenient for the representation of dissipation for the mass action law
is convenient for the representation of dissipation for the mass action law

This function may be considered as the sum of the reaction rates for deformed input stoichiometric coefficients 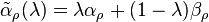 . For
. For 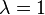 it is just the sum of the reaction rates. The function is convex because
it is just the sum of the reaction rates. The function is convex because 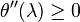 .
.
Direct calculation gives that according to the kinetic equations
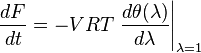
This is the general dissipation formula for the generalized mass action law.[25]
Convexity of gives the sufficient and necessary conditions for the proper dissipation inequality:
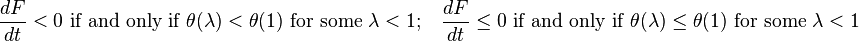
The semi-detailed balance condition can be transformed into identity  . Therefore, for the systems with semi-detailed balance
. Therefore, for the systems with semi-detailed balance 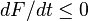 .[23]
.[23]
Detailed balance for systems with irreversible reactions
Detailed balance states that in equilibrium each elementary process is equilibrated by its reverse process and required reversibility of all elementary processes. For many real physico-chemical complex systems (e.g. homogeneous combustion, heterogeneous catalytic oxidation, most enzyme reactions etc.), detailed mechanisms include both reversible and irreversible reactions. If one represents irreversible reactions as limits of reversible steps, then it become obvious that not all reaction mechanisms with irreversible reactions can be obtained as limits of systems or reversible reactions with detailed balance. For example, the irreversible cycle cannot be obtained as such a limit but the reaction mechanism 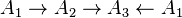 can.[26]
can.[26]
Gorban–Yablonsky theorem. A system of reactions with some irreversible reactions is a limit of systems with detailed balance when some constants tend to zero if and only if (i) the reversible part of this system satisfies the principle of detailed balance and (ii) the convex hull of the stoichiometric vectors of the irreversible reactions has empty intersection with the linear span of the stoichiometric vectors of the reversible reactions.[21] Physically, the last condition means that the irreversible reactions cannot be included in oriented cyclic pathways.
See also
- T-symmetry
- Microscopic reversibility
- Master equation
- Balance equation
- Gibbs sampling
- Metropolis–Hastings algorithm
- Atomic spectral line (deduction of the Einstein coefficients)
- Random walks on graphs
References
- ↑ 1.0 1.1 1.2 Boltzmann, L. (1964), Lectures on gas theory, Berkeley, CA, USA: U. of California Press.
- ↑ 2.0 2.1 Tolman, R. C. (1938). The Principles of Statistical Mechanics. Oxford University Press, London, UK.
- ↑ Maxwell, J.C. (1867), On the dynamical theory of gases, Philosl Trans R Soc London, 157 , pp. 49–88
- ↑ Einstein, A. (1916). Strahlungs-Emission und -Absorption nach der Quantentheorie [=Emission and absorption of radiation in quantum theory], Verhandlungen der Deutschen Physikalischen Gesellschaft 18 (13/14). Braunschweig: Vieweg, 318–323. See also: A. Einstein (1917). Zur Quantentheorie der Strahlung [=On the quantum theory of radiation], Physikalische Zeitschrift 18 (1917), 121–128. English translation: D. ter Haar (1967): The Old Quantum Theory. Pergamon Press, pp. 167–183.
- ↑ Wegscheider, R. (1901) Über simultane Gleichgewichte und die Beziehungen zwischen Thermodynamik und Reactionskinetik homogener Systeme, Monatshefte für Chemie / Chemical Monthly 32(8), 849–906.
- ↑ 6.0 6.1 Onsager, L. (1931), Reciprocal relations in irreversible processes. I, Phys. Rev. 37, 405–426; II 38, 2265–2279
- ↑ Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. (1953). "Equations of State Calculations by Fast Computing Machines". Journal of Chemical Physics 21 (6): 1087–1092. Bibcode:1953JChPh..21.1087M. doi:10.1063/1.1699114.
- ↑ van Kampen, N.G. "Stochastic Processes in Physics and Chemistry", Elsevier Science (1992).
- ↑ 9.0 9.1 9.2 Yablonskii, G.S., Bykov, V.I., Gorban, A.N., Elokhin, V.I. (1991), Kinetic Models of Catalytic Reactions, Amsterdam, The Netherlands: Elsevier.
- ↑ Lifshitz, E. M.; and Pitaevskii, L. P. (1981). Physical kinetics. London: Pergamon. ISBN 978-0-08-026480-6. Vol. 10 of the Course of Theoretical Physics(3rd Ed).
- ↑ 11.0 11.1 11.2 Gorban, A.N. (2014),Detailed balance in micro- and macrokinetics and micro-distinguishability of macro-processes, Results in Physics 4, 142–147
- ↑ Joshi, B. (2013), Deterministic detailed balance in chemical reaction networks is sufficient but not necessary for stochastic detailed balance, arXiv:1312.4196 [math.PR].
- ↑ 13.0 13.1 O'Hagan, Anthony; Forster, Jonathan (2004). "Section 10.3". Kendall's Advanced Theory of Statistics, Volume 2B: Bayesian Inference. New York: Oxford University Press. p. 263. ISBN 0-340-80752-0.
- ↑ Volpert, A.I., Khudyaev, S.I. (1985), Analysis in classes of discontinuous functions and equations of mathematical physics. Dordrecht, The Netherlands: Nijoff. (Translation from the 1st Russian ed., Moscow, Nauka publ., 1975.)
- ↑ Schuster, S., Schuster R. (1989). A generalization of Wegscheider's condition. Implications for properties of steady states and for quasi-steady-state approximation. J. Math. Chem, 3 (1), 25–42.
- ↑ Lorentz H.-A. (1887) Über das Gleichgewicht der lebendigen Kraft unter Gasmolekülen. Sitzungsberichte der Kaiserlichen Akademie der Wissenschaften in Wien. 95 (2), 115–152.
- ↑ 17.0 17.1 Boltzmann L. (1887) Neuer Beweis zweier Sätze über das Wärmegleichgewicht unter mehratomigen Gasmolekülen. Sitzungsberichte der Kaiserlichen Akademie der Wissenschaften in Wien. 95 (2), 153–164.
- ↑ Shannon, C.E. (1948) A Mathematical Theory of Communication, Bell System Technical Journal, Vol. 27, pp. 379–423, 623–656.
- ↑ Hugh Everett Theory of the Universal Wavefunction, Thesis, Princeton University, (1956, 1973), Appendix I, pp 121 ff. In his thesis, Everett used the term "detailed balance" unconventionally, instead of balance equation
- ↑ Cercignani, C. and Lampis, M. (1981). On the H-theorem for polyatomic gases, Journal of Statistical Physics, V. 26 (4), 795–801.
- ↑ 21.0 21.1 21.2 Gorban, A.N, Yablonsky, G.S. (2011) Extended detailed balance for systems with irreversible reactions, Chemical Engineering Science 66, 5388–5399.
- ↑ Colquhoun, D., Dowsland, K.A., Beato, M., and Plested, A.J.R. (2004) How to Impose Microscopic Reversibility in Complex Reaction Mechanisms, Biophysical Journal 86, June 2004, 3510–3518
- ↑ 23.0 23.1 Horn, F., Jackson, R. (1972) General mass action kinetics. Arch. Ration. Mech. Anal. 47, 87–116.
- ↑ Stueckelberg, E.C.G. (1952) Theoreme H et unitarite de S. Helv. Phys. Acta 25, 577–-580
- ↑ 25.0 25.1 25.2 Gorban, A.N., Shahzad, M. (2011) The Michaelis–Menten–Stueckelberg Theorem. Entropy 13, no. 5, 966–1019.
- ↑ Chu, Ch. (1971), Gas absorption accompanied by a system of first-order reactions, Chem. Eng. Sci. 26(3), 305–312.