Coupled cluster
Coupled cluster (CC) is a numerical technique used for describing many-body systems. Its most common use is as one of several post-Hartree–Fock ab initio quantum chemistry methods in the field of computational chemistry. It essentially takes the basic Hartree–Fock molecular orbital method and constructs multi-electron wavefunctions using the exponential cluster operator to account for electron correlation. Some of the most accurate calculations for small to medium sized molecules use this method.[1][2][3]
The method was initially developed by Fritz Coester and Hermann Kümmel in the 1950s for studying nuclear physics phenomena, but became more frequently used when in 1966 Jiři Čížek (and later together with Josef Paldus) reformulated the method for electron correlation in atoms and molecules. It is now one of the most prevalent methods in quantum chemistry that includes electronic correlation. CC theory is simply the perturbative variant of the Many Electron Theory (MET) of Oktay Sinanoğlu, which is the exact (and variational) solution of the many electron problem, so it was also called "Coupled Pair MET (CPMET)". J. Čížek used the correlation function of MET and used Goldstone type perturbation theory to get the energy expression while original MET was completely variational. Čížek first developed the Linear-CPMET and then generalized it to full CPMET in the same paper in 1966. He then also performed an application of it on benzene molecule with O. Sinanoğlu in the same year. Because MET is somewhat difficult to perform computationally, CC is simpler and thus, in today's computational chemistry, CC is the best variant of MET and gives highly accurate results in comparison to experiments.[4][5][6]
Wavefunction ansatz
Coupled-cluster theory provides the exact solution to the time-independent Schrödinger equation

where 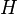 is the Hamiltonian of the system,
is the Hamiltonian of the system, 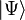 the exact wavefunction, and E the exact energy of the ground state. Coupled-cluster theory can also be used to obtain solutions for excited states using, for example, linear-response,[7] equation-of-motion,[8] state-universal multi-reference coupled cluster,[9] or valence-universal multi-reference coupled cluster[10] approaches.
the exact wavefunction, and E the exact energy of the ground state. Coupled-cluster theory can also be used to obtain solutions for excited states using, for example, linear-response,[7] equation-of-motion,[8] state-universal multi-reference coupled cluster,[9] or valence-universal multi-reference coupled cluster[10] approaches.
The wavefunction of the coupled-cluster theory is written as an exponential ansatz:
 ,
,
where 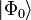 , the reference wave function, which is typically a Slater determinant constructed from Hartree–Fock molecular orbitals, though other wave functions such as Configuration interaction, Multi-configurational self-consistent field, or Brueckner orbitals can also be used.
, the reference wave function, which is typically a Slater determinant constructed from Hartree–Fock molecular orbitals, though other wave functions such as Configuration interaction, Multi-configurational self-consistent field, or Brueckner orbitals can also be used. 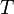 is the cluster operator which, when acting on , produces a linear combination of excited determinants from the reference wave function (see section below for greater detail).
is the cluster operator which, when acting on , produces a linear combination of excited determinants from the reference wave function (see section below for greater detail).
The choice of the exponential ansatz is opportune because (unlike other ansatzes, for example, configuration interaction) it guarantees the size extensivity of the solution. Size consistency in CC theory, however, depends on the size consistency of the reference wave function.
A criticism of the method is that the conventional implementation employing the similarity-transformed Hamiltonian (see below) is not variational, though there are bi-variational and quasi-variational approaches that have been developed since the first implementations of the theory. While the above ansatz for the wave function itself has no natural truncation, however, for other properties, such as energy, there is a natural truncation when examining expectation values, which has its basis in the linked- and connected-cluster theorems, and thus does not suffer from issues such as lack of size extensivity, like the variational configuration interaction.
Cluster operator
The cluster operator is written in the form,
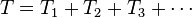 ,
,
where 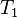 is the operator of all single excitations,
is the operator of all single excitations, 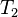 is the operator of all double excitations and so forth. In the formalism of second quantization these excitation operators are expressed as
is the operator of all double excitations and so forth. In the formalism of second quantization these excitation operators are expressed as
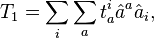
and for the general n-fold cluster operator
In the above formulae  and
and  denote the creation and annihilation operators respectively and i, j stand for occupied (hole) and a, b for unoccupied (particle) orbitals (states). The creation and annihilation operators in the coupled cluster terms above are written in canonical form, where each term is in the normal order form, with respect to the Fermi vacuum, . Being the one-particle cluster operator and the two-particle cluster operator, and convert the reference function into a linear combination of the singly and doubly excited Slater determinants, respectively, if applied without the exponential (such as in CI where a linear excitation operator is applied to the wave function). Applying the exponential cluster operator to the wave function, one can then generate more than doubly excited determinants due to the various powers of and that appear in the resulting expressions (see below). Solving for the unknown coefficients
denote the creation and annihilation operators respectively and i, j stand for occupied (hole) and a, b for unoccupied (particle) orbitals (states). The creation and annihilation operators in the coupled cluster terms above are written in canonical form, where each term is in the normal order form, with respect to the Fermi vacuum, . Being the one-particle cluster operator and the two-particle cluster operator, and convert the reference function into a linear combination of the singly and doubly excited Slater determinants, respectively, if applied without the exponential (such as in CI where a linear excitation operator is applied to the wave function). Applying the exponential cluster operator to the wave function, one can then generate more than doubly excited determinants due to the various powers of and that appear in the resulting expressions (see below). Solving for the unknown coefficients 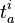 and
and 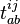 is necessary for finding the approximate solution .
is necessary for finding the approximate solution .
The exponential operator 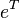 may be expanded as a Taylor series and if we consider only the and cluster operators of , we can write:
may be expanded as a Taylor series and if we consider only the and cluster operators of , we can write:
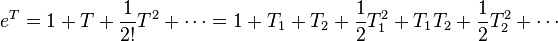
Though this series is finite in practice because the number of occupied molecular orbitals is finite, as is the number of excitations, it is still very large, to the extent that even modern day massively parallel computers are inadequate, except for problems of a dozen or so electrons and very small basis sets, when considering all contributions to the cluster operator and not just and . Often, as was done above, the cluster operator includes only singles and doubles (see CCSD below) as this offers a computationally affordable method that performs better than MP2 and CISD, but is not very accurate usually. For accurate results some form of triples (approximate or full) are needed, even near the equilibrium geometry (in the Franck-Condon region), and especially when breaking single-bonds or describing diradical species (these latter examples are often what is referred to as multi-reference problems, since more than one determinant has a significant contribution to the resulting wave function). For double bond breaking, and more complicated problems in chemistry, quadruple excitations often become important as well, though usually they are small for most problems, and as such, the contribution of 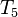 ,
, 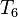 etc. to the operator is typically small. Furthermore, if the highest excitation level in the operator is n,
etc. to the operator is typically small. Furthermore, if the highest excitation level in the operator is n,

then Slater determinants for an N-electron system excited more than n (< N) times may still contribute to the coupled cluster wave function because of the non-linear nature of the exponential ansatz, and therefore, coupled cluster terminated at 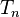 usually recovers more correlation energy than CI with maximum n excitations.
usually recovers more correlation energy than CI with maximum n excitations.
Coupled-cluster equations
The Schrödinger equation can be written, using the coupled-cluster wave function, as
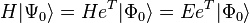
where there are a total of q coefficients (t-amplitudes) to solve for. To obtain the q equations, first, we multiply the above Schrödinger equation on the left by  and then project onto the entire set of up to m-tuply excited determinants, where m is the highest order excitation included in , that can be constructed from the reference wave function , denoted by
and then project onto the entire set of up to m-tuply excited determinants, where m is the highest order excitation included in , that can be constructed from the reference wave function , denoted by 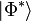 , and individually,
, and individually,  are singly excited determinants where the electron in orbital i has been excited to orbital a;
are singly excited determinants where the electron in orbital i has been excited to orbital a; 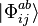 are doubly excited determinants where the electron in orbital i has been excited to orbital a and the electron in orbital j has been excited to orbital b, etc. In this way we generate a set of coupled energy-independent non-linear algebraic equations needed to determine the t-amplitudes.
are doubly excited determinants where the electron in orbital i has been excited to orbital a and the electron in orbital j has been excited to orbital b, etc. In this way we generate a set of coupled energy-independent non-linear algebraic equations needed to determine the t-amplitudes.
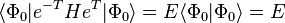
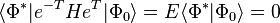 ,
,
(note, we have made use of 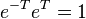 , the identity operator, and we are also assuming that we are using orthogonal orbitals, though this does not necessarily have to be true, e.g., valence bond orbitals, and in such cases the last set of equations are not necessarily equal to zero) the latter being the equations to be solved and the former the equation for the evaluation of the energy.
, the identity operator, and we are also assuming that we are using orthogonal orbitals, though this does not necessarily have to be true, e.g., valence bond orbitals, and in such cases the last set of equations are not necessarily equal to zero) the latter being the equations to be solved and the former the equation for the evaluation of the energy.
Considering the basic CCSD method:
 ,
, ,
, ,
,
in which the similarity transformed Hamiltonian, 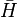 , can be explicitly written down using Hadamard's formula in Lie algebra, also called Hadamard's lemma (see also Baker–Campbell–Hausdorff formula (BCH formula), though note they are different, in that Hadamard's formula is a lemma of the BCH formula):
, can be explicitly written down using Hadamard's formula in Lie algebra, also called Hadamard's lemma (see also Baker–Campbell–Hausdorff formula (BCH formula), though note they are different, in that Hadamard's formula is a lemma of the BCH formula):
![\bar{H} = e^{-T} H e^{T} = H + [H,T] + (1/2)[[H,T],T] + ... = (He^{T})_{C}](../I/m/8ec8022b8cd9ebd5d3a6d904cbbe176b.png) .
.
The subscript C designates the connected part of the corresponding operator expression.
The resulting similarity transformed Hamiltonian is non-Hermitian, resulting in different left- and right-handed vectors (wave functions) for the same state of interest (this is what is often referred to in coupled cluster theory as the biorthogonality of the solution, or wave function, though it also applies to other non-Hermitian theories as well). The resulting equations are a set of non-linear equations which are solved in an iterative manner. Standard quantum chemistry packages (GAMESS (US), NWChem, ACES II, etc.) solve the coupled cluster equations using the Jacobi method and direct inversion of the iterative subspace (DIIS) extrapolation of the t-amplitudes to accelerate convergence.
Types of coupled-cluster methods
The classification of traditional coupled-cluster methods rests on the highest number of excitations allowed in the definition of . The abbreviations for coupled-cluster methods usually begin with the letters "CC" (for coupled cluster) followed by
- S - for single excitations (shortened to singles in coupled-cluster terminology)
- D - for double excitations (doubles)
- T - for triple excitations (triples)
- Q - for quadruple excitations (quadruples)
Thus, the operator in CCSDT has the form

Terms in round brackets indicate that these terms are calculated based on perturbation theory. For example, the CCSD(T) method means:
- Coupled cluster with a full treatment singles and doubles.
- An estimate to the connected triples contribution is calculated non-iteratively using Many-Body Perturbation Theory arguments.
General description of the theory
The complexity of equations and the corresponding computer codes, as well as the cost of the computation increases sharply with the highest level of excitation. For many applications CCSD, while relatively inexpensive, does not provide sufficient accuracy except for the smallest systems (approximately 2 to 4 electrons), and often an approximate treatment of triples is needed. The most well known coupled cluster method that provides an estimate of connected triples is CCSD(T), which provides a good description of closed-shell molecules near the equilibrium geometry, but breaks down in more complicated situations such as bond breaking and diradicals. Another popular method that makes up for the failings of the standard CCSD(T) approach is CR-CC(2,3), where the triples contribution to the energy is computed from the difference between the exact solution and the CCSD energy, and is not based on perturbation theory arguments. More complicated coupled-cluster methods such as CCSDT and CCSDTQ are used only for high-accuracy calculations of small molecules. The inclusion of all n levels of excitation for the n-electron system gives the exact solution of the Schrödinger equation within the given basis set, within the Born–Oppenheimer approximation (although schemes have also been drawn up to work without the BO approximation[11][12]).
One possible improvement to the standard coupled-cluster approach is to add terms linear in the interelectronic distances through methods such as CCSD-R12. This improves the treatment of dynamical electron correlation by satisfying the Kato cusp condition and accelerates convergence with respect to the orbital basis set. Unfortunately, R12 methods invoke the resolution of the identity which requires a relatively large basis set in order to be a good approximation.
The coupled-cluster method described above is also known as the single-reference (SR) coupled-cluster method because the exponential ansatz involves only one reference function . The standard generalizations of the SR-CC method are the multi-reference (MR) approaches: state-universal coupled cluster (also known as Hilbert space coupled cluster), valence-universal coupled cluster (or Fock space coupled cluster) and state-selective coupled cluster (or state-specific coupled cluster).
Historical accounts
In the first reference below, Kümmel comments:
- Considering the fact that the CC method was well understood around the late fifties it looks strange that nothing happened with it until 1966, as Jiři Čížek published his first paper on a quantum chemistry problem. He had looked into the 1957 and 1960 papers published in Nuclear Physics by Fritz and myself. I always found it quite remarkable that a quantum chemist would open an issue of a nuclear physics journal. I myself at the time had almost gave up the CC method as not tractable and, of course, I never looked into the quantum chemistry journals. The result was that I learnt about Jiři's work as late as in the early seventies, when he sent me a big parcel with reprints of the many papers he and Joe Paldus had written until then.
Josef Paldus also wrote his first hand account of the origins of coupled-cluster theory, its implementation, and exploitation in electronic wave function determination; his account is primarily about the making of coupled-cluster theory rather than about the theory itself.[13]
Relation to other theories
Configuration Interaction
The Cj excitation operators defining the CI expansion of an N-electron system for the wave function 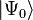 ,
,
 ,
,
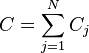 ,
,
are related to the cluster operators , since in the limit of including up to 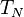 in the cluster operator the CC theory must be equal to full CI, we obtain the following relationships[14][15]
in the cluster operator the CC theory must be equal to full CI, we obtain the following relationships[14][15]
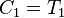 ,
,
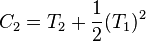 ,
,
 ,
,
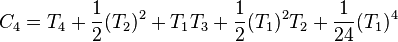 ,
,
etc. For general relationships see J. Paldus, in Methods in Computational Molecular Physics, Vol. 293 of Nato Advanced Study Institute Series B: Physics, edited by S. Wilson and G.H.F. Diercksen (Plenum, New York, 1992), pp. 99–194.
Symmetry Adapted Cluster
The Symmetry adapted cluster (SAC)[16][17] approach determines the (spin and) symmetry adapted cluster operator

by solving the following system of energy dependent equations,
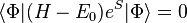 ,
,
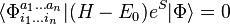 ,
,
 ,
, 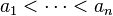 ,
,  ,
,
where 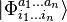 are the n-tuply excited determinants relative to
are the n-tuply excited determinants relative to  (usually they are the spin- and symmetry-adapted configuration state functions, in practical implementations), and
(usually they are the spin- and symmetry-adapted configuration state functions, in practical implementations), and 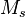 is the highest-order of excitation included in the SAC operator. If all of the nonlinear terms in
is the highest-order of excitation included in the SAC operator. If all of the nonlinear terms in 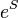 are included then the SAC equations become equivalent to the standard coupled-cluster equations of Jiři Čížek. This is due to the cancellation of the energy-dependent terms with the disconnected terms contributing to the product of
are included then the SAC equations become equivalent to the standard coupled-cluster equations of Jiři Čížek. This is due to the cancellation of the energy-dependent terms with the disconnected terms contributing to the product of 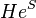 , resulting in the same set of nonlinear energy-independent equations. Typically, all nonlinear terms, except
, resulting in the same set of nonlinear energy-independent equations. Typically, all nonlinear terms, except  are dropped, as higher-order nonlinear terms are usually small.[18]
are dropped, as higher-order nonlinear terms are usually small.[18]
See also
- Quantum chemistry computer programs
References
- ↑ Kümmel, H. G. (2002). "A biography of the coupled cluster method". In Bishop, R. F.; Brandes, T.; Gernoth, K. A.; Walet, N. R.; Xian, Y. Recent progress in many-body theories Proceedings of the 11th international conference. Singapore: World Scientific Publishing. pp. 334–348. ISBN 978-981-02-4888-8.
- ↑ Cramer, Christopher J. (2002). Essentials of Computational Chemistry. Chichester: John Wiley & Sons, Ltd. pp. 191–232. ISBN 0-471-48552-7.
- ↑ Shavitt, Isaiah; Bartlett, Rodney J. (2009). Many-Body Methods in Chemistry and Physics: MBPT and Coupled-Cluster Theory. Cambridge University Press. ISBN 978-0-521-81832-2.
- ↑ Čížek, Jiří (1966). "On the Correlation Problem in Atomic and Molecular Systems. Calculation of Wavefunction Components in Ursell-Type Expansion Using Quantum-Field Theoretical Methods". The Journal of Chemical Physics 45 (11): 4256. Bibcode:1966JChPh..45.4256C. doi:10.1063/1.1727484.
- ↑ Sinanoğlu, O.; Brueckner, K. (1971). Three approaches to electron correlation in atoms. Yale Univ. Press. ISBN 0-300-01147-4. and references therein
- ↑ Si̇nanoğlu, Oktay (1962). "Many-Electron Theory of Atoms and Molecules. I. Shells, Electron Pairs vs Many-Electron Correlations". The Journal of Chemical Physics 36 (3): 706. Bibcode:1962JChPh..36..706S. doi:10.1063/1.1732596.
- ↑ Monkhorst, H.J. (1977). "Calculation of properties with the coupled-cluster method". International Journal of Quantum Chemistry. 12, S11: 421. doi:10.1002/qua.560120850.
- ↑ Stanton, John F.; Bartlett, Rodney J. (1993). "The equation of motion coupled-cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties". The Journal of Chemical Physics 98 (9): 7029. Bibcode:1993JChPh..98.7029S. doi:10.1063/1.464746.
- ↑ Jeziorski, B.; Monkhorst, H. (1981). "Coupled-cluster method for multideterminantal reference states". Physical Review A 24 (4): 1668. Bibcode:1981PhRvA..24.1668J. doi:10.1103/PhysRevA.24.1668.
- ↑ Lindgren, D.; Mukherjee, Debashis (1987). "On the connectivity criteria in the open-shell coupled-cluster theory for general model spaces". Physics Reports 151 (2): 93. Bibcode:1987PhR...151...93L. doi:10.1016/0370-1573(87)90073-1.
- ↑ Monkhorst, Hendrik J (1987). "Chemical physics without the Born-Oppenheimer approximation: The molecular coupled-cluster method". Physical Review A 36 (4): 1544–1561. Bibcode:1987PhRvA..36.1544M. doi:10.1103/PhysRevA.36.1544. PMID 9899035.
- ↑ Nakai, Hiromi; Sodeyama, Keitaro (2003). "Many-body effects in nonadiabatic molecular theory for simultaneous determination of nuclear and electronic wave functions: Ab initio NOMO/MBPT and CC methods". The Journal of Chemical Physics 118 (3): 1119. Bibcode:2003JChPh.118.1119N. doi:10.1063/1.1528951.
- ↑ Paldus, J. (2005). "The beginnings of coupled-cluster theory: an eyewitness account". In Dykstra, C. Theory and Applications of Computational Chemistry: The First Forty Years. Elsivier B.V. p. 115.
- ↑ Paldus, J (1981). Diagrammatic Methods for Many-Fermion Systems (Lecture Notes ed.). University of Nijmegen, Njimegen, The Netherlands.
- ↑ Bartlett, R.J.; Dykstra, C.E.; Paldus, J. (1984). Dykstra, C.E., ed. Advanced Theories and Computational Approaches to the Electronic Structure of Molecules. p. 127.
- ↑ Nakatsuji, H.; Hirao, K. (1977). "Cluster expansion of the wavefunction. Pseduo-orbital theory applied to spin correlation". Chemical Physics Letters 47 (3): 569. Bibcode:1977CPL....47..569N. doi:10.1016/0009-2614(77)85042-2.
- ↑ Nakatsuji, H.; Hirao, K. (1978). "Cluster expansion of the wavefunction. Symmetry‐adapted‐cluster expansion, its variational determination, and extension of open‐shell orbital theory". Journal of Chemical Physics 68 (5): 2053. Bibcode:1978JChPh..68.2053N. doi:10.1063/1.436028.
- ↑ Ohtsuka, Y.; Piecuch, P.; Gour, J.R.; Ehara, M.; Nakatsuji, H. (2007). "Active-space symmetry-adapted-cluster configuration-interaction and equation-of-motion coupled-cluster methods for high accuracy calculations of potential energy surfaces of radicals". Journal of Chemical Physics 126 (16): 164111. Bibcode:2007JChPh.126p4111O. doi:10.1063/1.2723121. PMID 17477593.