Comparative genomic hybridization
Comparative genomic hybridization is a molecular cytogenetic method for analysing copy number variations (CNVs) relative to ploidy level in the DNA of a test sample compared to a reference sample, without the need for culturing cells. The aim of this technique is to quickly and efficiently compare two genomic DNA samples arising from two sources, which are most often closely related, because it is suspected that they contain differences in terms of either gains or losses of either whole chromosomes or subchromosomal regions (a portion of a whole chromosome). This technique was originally developed for the evaluation of the differences between the chromosomal complements of solid tumor and normal tissue,[1] and has an improved resoIution of 5-10 megabases compared to the more traditional cytogenetic analysis techniques of giemsa banding and fluorescence in situ hybridization (FISH) which are limited by the resolution of the microscope utilized.[2][3]
This is achieved through the use of competitive fluorescence in situ hybridization. In short, this involves the isolation of DNA from the two sources to be compared, most commonly a test and reference source, independent labelling of each DNA sample with a different fluorophores (fluorescent molecules) of different colours (usually red and green), denaturation of the DNA so that it is single stranded, and the hybridization of the two resultant samples in a 1:1 ratio to a normal metaphase spread of chromosomes, to which the labelled DNA samples will bind at their locus of origin. Using a fluorescence microscope and computer software, the differentially coloured fluorescent signals are then compared along the length of each chromosome for identification of chromosomal differences between the two sources. A higher intensity of the test sample colour in a specific region of a chromosome indicates the gain of material of that region in the corresponding source sample, while a higher intensity of the reference sample colour indicates the loss of material in the test sample in that specific region. A neutral colour (yellow when the fluorophore labels are red and green) indicates no difference between the two samples in that location.[2][3]
CGH is only able to detect unbalanced chromosomal abnormalities. This is because balanced chromosomal abnormalities such as reciprocal translocations, inversions or ring chromosomes do not affect copy number, which is what is detected by CGH technologies. CGH does, however, allow for the exploration of all 46 human chromosomes in single test and the discovery of deletions and duplications, even on the microscopic scale which may lead to the identification of candidate genes to be further explored by other cytological techniques.[2][4]
Through the use of DNA microarrays in conjunction with CGH techniques, the more specific form of array CGH (aCGH) has been developed, allowing for a locus-by-locus measure of CNV with increased resolution as low as 100 kilobases.[5][6] This improved technique allows for the aetiology of known and unknown conditions to be discovered.
History
The motivation underlying the development of CGH stemmed from the fact that the available forms of cytogenetic analysis at the time (giemsa banding and FISH) were limited in their potential resolution by the microscopes necessary for interpretation of the results they provided. Furthermore, giemsa banding interpretation has the potential to be ambiguous and therefore has lowered reliability, and both techniques require high labour inputs which limits the loci which may be examined.[5]
The first report of CGH analysis was by Kallioniemi and colleagues in 1992 at the University of California, San Francisco, who utilised CGH in the analysis of solid tumors. They achieved this by the direct application of the technique to both breast cancer cell lines and primary bladder tumors in order to establish complete copy number karyotypes for the cells. They were able to identify 16 different regions of amplification, many of which were novel discoveries.[1]
Soon after in 1993, du Manoir et al. reported virtually the same methodology. The authors painted a series of individual human chromosomes from a DNA library with two different fluorophores in different proportions to test the technique, and also applied CGH to genomic DNA from patients affected with either Downs syndrome or T-cell prolymphocytic leukemia as well as cells of a renal papillary carcinoma cell line. It was concluded that the fluorescence ratios obtained were accurate and that differences between genomic DNA from different cell types were detectable, and therefore that CGH was a highly useful cytogenetic analysis tool.[7]
Initially, the widespread use of CGH technology was difficult, as protocols were not uniform and therefore inconsistencies arose, especially due to uncertainties in the interpretation of data.[3] However, in 1994 a review was published which described an easily understood protocol in detail[8] and the image analysis software was made available commercially, which allowed CGH to be utilised all around the world.[3] As new techniques such as microdissection and degenerate oligonucleotide primed polymerase chain reaction (DOP-PCR) became available for the generation of DNA products, it was possible to apply the concept of CGH to smaller chromosomal abnormalities, and thus the resolution of CGH was improved.[3]
The implementation of array CGH, whereby DNA microarrays are used instead of the traditional metaphase chromosome preparation, was pioneered by Solinas-Tolodo et al. in 1997 using tumor cells[9] and Pinkel et al. in 1998 by use of breast cancer cells.[10] This was made possible by the Human Genome Project which generated a library of cloned DNA fragments with known locations throughout the human genome, with these fragments being used as probes on the DNA microarray.[11] Now probes of various origins such as cDNA, genomic PCR products and bacterial artificial chromosomes (BACs) can be used on DNA microarrays which may contain up to 2 million probes.[11] Array CGH is automated, allows greater resolution (down to 100 kb) than traditional CGH as the probes are far smaller than metaphase preparations, requires smaller amounts of DNA, can be targeted to specific chromosomal regions if required and is ordered and therefore faster to analyse, making it far more adaptable to diagnostic uses.[11][12]

Basic Methods
Metaphase Slide Preparation
The DNA on the slide is a reference sample, and is thus obtained from a karyotypically normal man or woman, though it is preferential to use female DNA as they possess two X chromosomes which contain far more genetic information than the male Y chromosome. Phytohaemagglutinin stimulated peripheral blood lymphocytes are used. 1mL of heparinised blood is added to 10ml of culture medium and incubated for 72 hours at 37 °C in an atmosphere of 5% CO_2. Colchicine is added to arrest the cells in mitosis, the cells are then harvested and treated with hypotonic potassium chloride and fixed in 3:1 methanol/acetic acid.[3]
One drop of the cell suspension should then be dropped onto an ethanol cleaned slide from a distance of about 30 cm, optimally this should be carried out at room temperature at humidity levels of 60-70%. Slides should be evaluated by visualisation using a phase contrast microscope, minimal cytoplasm should be observed and chromosomes should not be overlapping and be 400-550 bands long with no separated chromatids and finally should appear dark rather than shiny. Slides then need to be air dried overnight at room temperature, and any further storage should be in groups of four at -20 °C with either silica beads or nitrogen present to maintain dryness. Different donors should be tested as hybridization may be variable. Commercially available slides may be used, but should always be tested first.[3]
Isolation of DNA from Test Tissue and Reference Tissue
Standard phenol extraction is used to obtain DNA from test or reference (karyotypically normal individual) tissue, which involves the combination of Tris-Ethylenediaminetetraacetic acid and phenol with aqueous DNA in equal amounts. This is followed by separation by agitation and centrifugation, after which the aqueous layer is removed and further treated using ether and finally ethanol precipitation is used to concentrate the DNA.[3]
May be completed using DNA isolation kits available commercially which are based on affinity columns.[3]
Preferentially, DNA should be extracted from fresh or frozen tissue as this will be of the highest quality, though it is now possible to use archival material which is fornaline fixed or paraffin wax embedded, provided the appropriate procedures are followed. 0.5-1 µg of DNA is sufficient for the CGH experiment, though if the desired amount is not obtained DOP-PCR may be applied to amplify the DNA, however it in this case it is important to apply DOP-PCR to both the test and reference DNA samples to improve reliability.[3]
DNA Labelling
Nick translation is used to label the DNA and involves cutting DNA and substituting nucleotides labelled with fluorophores (direct labelling) or biotin or oxigenin to have fluophore conjugated antibodies added later (indirect labelling). It is then important to check fragment lengths of both test and reference DNA by gel electrophoresis, as they should be within the range of 500kb-1500kb for optimum hybridization.[3]
Blocking
Unlabelled Life Technologies Corporation's Cot-1 DNA® (placental DNA enriched with repetitive sequences of length 50bp-100bp)is added to block normal repetitive DNA sequences, particularly at centromeres and telomeres, as if these sequences are detected, they may reduce the fluorescence ratio and cause gains or losses to escape detection.[3]
Hybridization
8-12µl of each of labelled test and labelled reference DNA are mixed and 40 µg Cot-1 DNA® is added, then precipitated and subsequently dissolved in 6µl of hybridization mix, which contains 50% formamide to decrease DNA melting temperature and 10% dextran sulphate to increase the effective probe concentration in a saline sodium citrate (SSC) solution at a pH of 7.0.[3]
Denaturation of the slide and probes are carried out separately. The slide is submerged in 70% formamide/2xSSC for 5–10 minutes at 72 °C, while the probes are denatured by immersion in a water bath of 80 °C for 10 minutes and are immediately added to the metaphase slide preparation. This reaction is then covered with a coverslip and left for two to four days in a humid chamber at 40 °C.[3]
The coverslip is then removed and 5 minute washes are applied, three using 2xSSC at room temperature, one at 45 °C with 0.1xSSC and one using TNT at room temperature. The reaction is then preincubated for 10 minutes then followed by a 60 minute, 37 °C incubation, three more 5 minute washes with TNT then one with 2xSSC at room temperature. The slide is then dried using an ethanol series of 70%/96%/100% before counterstaining with DAPI (0.35 μg/ml), for chromosome identification, and sealing with a coverslip.[3]
Fluorescence Visualisation and Imaging
A fluorescence microscope with the appropriate filters for the DAPI stain as well as the two fluorophores utilised is required for visualisation, and these filters should also minimise the crosstalk between the fluorophores, such as narrow band pass filters. The microscope must provide uniform illumination without chromatic variation, be appropriately aligned and have a “plan” type of objective which is apochromatic and give a magnification of x63 or x100.[3]
The image should be recorded using a camera with spatial resolution at least 0.1 µm at the specimen level and give an image of at least 600x600 pixels. The camera must also be able to integrate the image for at least 5 to 10 seconds, with a minimum photometric resolution of 8 bit.[3]
Dedicated CGH software is commercially available for the image processing step, and is required to subtract background noise, remove and segment materials not of chromosomal origin, normalize the fluorescence ratio, carry out interactive karyotyping and chromosome scaling to standard length. A “relative copy number karyotype” which presents chromosomal areas of deletions or amplifications is generated by averaging the ratios of a number of high quality metaphases and plotting them along an ideogram, a diagram identifying chromosomes based on banding patterns. Interpretation of the ratio profiles is conducted either using fixed or statistical thresholds (confidence intervals). When using confidence intervals, gains or losses are identified when 95% of the fluorescence ratio does not contain 1.0.[3]
Extra Notes
Extreme care must be taken to avoid contamination of any step involving DNA, especially with the test DNA as contamination of the sample with normal DNA will skew results closer to 1.0, thus abnormalities may go undetected. FISH, PCR and flow cytometry experiments may be employed to confirm results.[5][13]
Array Comparative Genomic Hybridization
Array comparative genomic hybridization (also microarray-based comparative genomic hybridization, matrix CGH, array CGH, aCGH) is a molecular cytogenetic technique for the detection of chromosomal copy number changes on a genome wide and high-resolution scale.[14] Array CGH compares the patient’s genome against a reference genome and identifies differences between the two genomes, and hence locates regions of genomic imbalances in the patient, utilizing the same principles of competitive fluorescence in situ hybridization as traditional CGH.
With the introduction of array CGH, the main limitation of conventional CGH, a low resolution, is overcome. In array CGH, the metaphase chromosomes are replaced by cloned DNA fragments (+100–200 kb) of which the exact chromosomal location is known. This allows the detection of aberrations in more detail and, moreover, makes it possible to map the changes directly onto the genomic sequence.[15]
Array CGH has proven to be a specific, sensitive, fast and highthroughput technique, with considerable advantages compared to other methods used for the analysis of DNA copy number changes making it more amenable to diagnostic applications. Using this method, copy number changes at a level of 5–10 kilobases of DNA sequences can be detected.[16] As of 2006, even high-resolution CGH (HR-CGH) arrays are accurate to detect structural variations (SV) at resolution of 200 bp.[17] This method allows one to identify new recurrent chromosome changes such as microdeletions and duplications in human conditions such as cancer and birth defects due to chromosome aberrations.
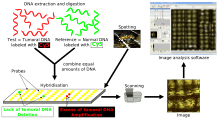
Methodology
Array CGH is based on the same principle as conventional CGH. In both techniques, DNA from a reference (or control) sample and DNA from a test (or patient) sample are differentially labelled with two different fluorophores and used as probes that are cohybridized competitively onto nucleic acid targets. In conventional CGH, the target is a reference metaphase spread. In array CGH, these targets can be genomic fragments cloned in a variety of vectors (such as BACs or plasmids), cDNAs, or oligonucleotides.[18]
Figure 2.[15] is a schematic overview of the array CGH technique. DNA from the sample to be tested is labeled with a green fluorophore (Cyanine 3) and a reference DNA sample is labeled with red fluorophore (Cyanine 5). Equal quantities of the two DNA samples are mixed and cohybridized to a DNA microarray of several thousand evenly spaced cloned DNA fragments or oligonucleotides, which have been spotted in triplicate on the array. After hybridization, digital imaging systems are used to capture and quantify the relative fluorescence intensities of each of the hybridized fluorophores.[18] The resulting ratio of the fluorescence intensities is proportional to the ratio of the copy numbers of DNA sequences in the test and reference genomes. If the intensities of the flurochromes are equal on one probe, this region of the patient’s genome is interpreted as having equal quantity of DNA in the test and reference samples; if there is an altered Cy3:Cy5 ratio this indicates a loss or a gain of the patient DNA at that specific genomic region.[19]
Technological Approaches to Array CGH

Array CGH has been implemented using a wide variety of techniques. Therefore, some of the advantages and limitations of array CGH are dependent on the technique chosen. The initial approaches used arrays produced from large insert genomic DNA clones, such as BACs. The use of BACs provides sufficient intense signals to detect single-copy changes and to locate aberration boundaries accurately. However, initial DNA yields of isolated BAC clones are low and DNA amplification techniques are necessary. These techniques include ligation-mediated polymerase chain reaction (PCR), degenerate primer PCR using one or several sets of primers, and rolling circle amplification.[20] Arrays can also be constructed using cDNA. These arrays currently yield a high spatial resolution, but the number of cDNAs is limited by the genes that are encoded on the chromosomes, and their sensitivity is low due to cross-hybridization.[15] This results in the inability to detect single copy changes on a genome wide scale.[21] The latest approach is spotting the arrays with short oligonucleotides. The amount of oligos is almost infinite, and the processing is rapid, cost-effective, and easy. Although oligonucleotides do not have the sensitivity to detect single copy changes, averaging of ratios from oligos that map next to each other on the chromosome can compensate for the reduced sensitivity.[22] It is also possible to use arrays which have overlapping probes so that specific breakpoints may be uncovered.
Design Approaches
There are two approaches to the design of microarrays for CGH applications: whole genome and targeted.
Whole genome arrays are designed to cover the entire human genome. They often include clones that provide an extensive coverage across the genome; and arrays that have contiguous coverage, within the limits of the genome. Whole-genome arrays have been constructed mostly for research applications and have proven their outstanding worth in gene discovery. They are also very valuable in screening the genome for DNA gains and losses at an unprecedented resolution.[18]
Targeted arrays are designed for a specific region(s) of the genome for the purpose of evaluating that targeted segment. It may be designed to study a specific chromosome or chromosomal segment or to identify and evaluate specific DNA dosage abnormalities in individuals with suspected microdeletion syndromes or subtelomeric rearrangements. The crucial goal of a targeted microarray in medical practice is to provide clinically useful results for diagnosis, genetic counseling, prognosis, and clinical management of unbalanced cytogenetic abnormalities.[18]
Applications
Conventional CGH
Conventional CGH has been used mainly for the identification of chromosomal regions that are recurrently lost or gained in tumors, as well as for the diagnosis and prognosis of cancer.[23] This approach can also be used to study chromosomal aberrations in fetal and neonatal genomes. Furthermore, conventional CGH can be used in detecting chromosomal abnormalities and have been shown to be efficient in diagnosing complex abnormalities associated with human genetic disorders.[15]
CGH in cancer research
CGH data from several studies of the same tumor type show consistent patterns of non-random genetic aberrations.[24] Some of these changes appear to be common to various kinds of malignant tumors, while others are more tumor specific. For example, gains of chromosomal regions lq, 3q and 8q, as well as losses of 8p, 13q, 16q and 17p, are common to a number of tumor types, such as breast, ovarian, prostate, renal and bladder cancer (Figure. 3). Other alterations, such as 12p and Xp gains in testicular cancer, 13q gain 9q loss in bladder cancer, 14q loss in renal cancer and Xp loss in ovarian cancer are more specific, and might reflect the unique selection forces operating during cancer development in different organs.[24]
An overview of genome copy number profiles for the majority of cancer types is presented through the Progenetix project.[25][26]
Chromosomal Aberrations
Cri du Chat (CdC) is a syndrome caused by a partial deletion of the short arm of chromosome 5.[27] Several studies have shown that conventional CGH is suitable to detect the deletion, as well as more complex chromosomal alterations. For example, Levy et al. (2002) reported an infant with a cat-like cry, the hallmark of CdC, but having an indistinct karyotype. CGH analysis revealed a loss of chromosomal material from 5p15.3 confirming the diagnosis clinically. These results demonstrate that conventional CGH is a reliable technique in detecting structural aberrations and, in specific cases, may be more efficient in diagnosing complex abnormalities.[27]
Array CGH
Array CGH applications are mainly directed at detecting genomic abnormalities in cancer. However, array CGH is also suitable for the analysis of DNA copy number aberrations that cause human genetic disorders.[15] That is, array CGH is employed to uncover deletions, amplifications, breakpoints and ploidy abnormalities. Earlier diagnosis is of benefit to the patient as they may undergo appropriate treatments and counseling to improve their prognosis.[11]
Genomic abnormalities in cancer
Genetic alterations and rearrangements occur frequently in cancer and contribute to its pathogenesis. Detecting these aberrations by array CGH provides information on the locations of important cancer genes and can have clinical use in diagnosis, cancer classification and prognostification.[18] However, not all of the losses of genetic material are pathogenetic, since some DNA material is physiologically lost during the rearrangement of immunoglobulin subgenes.[28]
Array CGH may also be applied not only to the discovery of chromosomal abnormalities in cancer, but also to the monitoring of the progression of tumors. Differentiation between metastatic and mild lesions is also possible using FISH once the abnormalities have been identified by array CGH.[6][11]
The arrayMap web site [29] offers access to pre-processed copy number profiles as well as clinical annotations from some ten thousands of genomic arrays, as well as online visualisation tools and programmatic data access.
Submicroscopic aberrations
Prader–Willi syndrome (PWS) is a paternal structural abnormality involving 15q11-13, while a maternal aberration in the same region causes Angelman syndrome (AS). In both syndromes, the majority of cases (75%) are the result of a 3–5 Mb deletion of the PWS/AS critical region.[30] These small aberrations cannot be detected using cytogenetics or conventional CGH, but can be readily detected using array CGH. As a proof of principle Vissers et al. (2003) constructed a genome wide array with a 1 Mb resolution to screen three patients with known, FISH-confirmed microdeletion syndromes, including one with PWS. In all three cases, the abnormalities, ranging from 1.5 to 2.9Mb, were readily identified.[31] Thus, array CGH was demonstrated to be a specific and sensitive approach in detecting submicroscopic aberrations.
When using overlapping microarrays, it is also possible to uncover breakpoints involved in chromosomal aberrations.
Prenatal Genetic Diagnosis
Though not yet a widely employed technique, the use of array CGH as a tool for preimplantation genetic screening is becoming an increasingly popular concept. It has the potential to detect CNVs and aneuploidy in eggs, sperm or embryos which may contribute to failure of the embryo to successfully implant, miscarriage or conditions such as Down syndrome (trisomy 21). This makes array CGH a promising tool to reduce the incidence of life altering conditions and improve success rates of IVF attempts. The technique involves whole genome amplification from a single cell which is then used in the array CGH method. It may also be used in couples carrying chromosomal translocations such as balanced reciprocal translocations or Robertsonian translocations, which have the potential to cause chromosomal imbalances in their offspring.[13][32][33]
Limitations of CGH and array CGH
A main disadvantage of conventional CGH is its inability to detect structural chromosomal aberrations, without copy number changes, such as mosaicism, balanced chromosomal translocations, and inversions. CGH can also only detect gains and losses relative to the ploidy level.[34] In addition, chromosomal regions with short repetitive DNA sequences are highly variable between individuals and can interfere with CGH analysis.[15] Therefore, repetitive DNA regions like centromeres and telomeres need to be blocked with unlabeled repetitive DNA (e.g. Cot1 DNA) and/or can be omitted from screening.[35] Furthermore, the resolution of conventional CGH is a major practical problem that limits its clinical applications. Although CGH has proven to be a useful and reliable technique in the research and diagnostics of both cancer and human genetic disorders, the applications involve only gross abnormalities. Because of the limited resolution of metaphase chromosomes, aberrations smaller than 5–10 Mb cannot be detected using conventional CGH.[24] For the detection of such abnormalities, a high-resolution technique is required. Array CGH overcomes many of these limitations. Array CGH is characterized by a high resolution, its major advantage with respect to conventional CGH. The standard resolution varies between 1 and 5 Mb, but can be increased up to approximately 40 kb by supplementing the array with extra clones. However, as in conventional CGH, the main disadvantage of array CGH is its inability to detect aberrations that do not result in copy number changes and is limited in its ability to detect mosaicism.[15] The level of mosaicism that can be detected is dependent on the sensitivity and spatial resolution of the clones. At present, rearrangements present in approximately 50% of the cells is the detection limit. For the detection of such abnormalities, other techniques, such as SKY (Spectral karyotyping) or FISH have to still be used.[36] Another disadvantage is the lack of commercial availability of the arrays. Until now, BAC and cDNA clones can be obtained from, for example, the Sanger Center, BACPAC Resources, Invitrogen and Research Genetics. However, array preparation still needs to be performed by the investigators themselves.[15] Inconsistencies in visualization and imaging software as well as interpretation parameters also make it difficult for replications and comparisons to be made by different laboratory teams.[2][3]
See also
- Array based Comparative Genomic Hybridization
- Virtual Karyotype
- Cytogenetics
- Fluorescence in situ hybridisation
References
- ↑ 1.0 1.1 Kallioniemi A, Kallioniemi OP, Sudar Da, Rutovitz D, Gray JW, Waldman F, Pinkel D (1992) Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258:818-821.
- ↑ 2.0 2.1 2.2 2.3 Strachan T, Read AP (2010) Human Molecular Genetics: Garland Science.
- ↑ 3.0 3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 3.9 3.10 3.11 3.12 3.13 3.14 3.15 3.16 3.17 3.18 Weiss M, Hermsen M, Meijer G, Van Grieken N, Baak J, Kuipers E, Van Diest P (1999) Comparative genomic hybridization. Molecular Pathology 52:243-251.
- ↑ [3]
- ↑ 5.0 5.1 5.2 Pinkel D, Albertson DG (2005) Comparative genomic hybridization. Annu Rev Genomics Hum Genet 6:331-354.
- ↑ 6.0 6.1 de Ravel TJ, Devriendt K, Fryns J-P, Vermeesch JR (2007) What’s new in karyotyping? The move towards array comparative genomic hybridization (CGH). European journal of pediatrics 166:637-643.
- ↑ du Manoir S, Speicher MR, Joos S, Schröck E, Popp S, Döhner H, Kovacs G, Robert-Nicoud M, Lichter P, Cremer T (1993) Detection of complete and partial chromosome gains and losses by comparative genomic in situ hybridization. Human genetics 90:590-610.
- ↑ Kallioniemi OP, Kallioniemi A, Piper J, Isola J, Waldman FM, Gray JW, Pinkel D (1994) Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors. Genes, Chromosomes and Cancer 10:231-243.
- ↑ Solinas‐Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Döhner H, Cremer T, Lichter P (1997) Matrix‐based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes, Chromosomes and Cancer 20:399-407.
- ↑ Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo W-L, Chen C, Zhai Y (1998) High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nature genetics 20:207-211.
- ↑ 11.0 11.1 11.2 11.3 11.4 Marquis-Nicholson R, Aftimos S, Hayes I, George A, Love DR (2010) Array comparative genomic hybridization: a new tool in the diagnostic genetic armoury. NZ Med J 123:50-61.
- ↑ Inazawa J, Inoue J, Imoto I (2004) Comparative genomic hybridization (CGH)‐arrays pave the way for identification of novel cancer‐related genes. Cancer Science 95:559-563.
- ↑ 13.0 13.1 Evangelidou P, Alexandrou A, Moutafi M, Ioannides M, Antoniou P, Koumbaris G, Kallikas I, Velissariou V, Sismani C, Patsalis PC (2013) Implementation of High Resolution Whole Genome Array CGH in the Prenatal Clinical Setting: Advantages, Challenges, and Review of the Literature. BioMed Research International 2013.
- ↑ Pinkel D, Albertson DG (2005) Array comparative genomic hybridization and its applications in cancer. Nat Genet 37:11-17.
- ↑ 15.0 15.1 15.2 15.3 15.4 15.5 15.6 15.7 Oostlander AE, Meijer GA, Ylstra B (2004) Microarray-based comparative genomic hybridization and its applications in human genetics. Clin Genet 66:488-495.
- ↑ Ren H, Francis W, Boys A, Chueh AC, Wong N, La P, Wong LH, Ryan J, Slater HR, Choo KH (May 2005). "BAC-based PCR fragment microarray: high-resolution detection of chromosomal deletion and duplication breakpoints". Human Mutation 25 (5): 476–82. doi:10.1002/humu.20164. PMID 15832308.
- ↑ Urban AE, Korbel JO, Selzer R, Richmond T, Hacker A, Popescu GV, Cubells JF, Green R, Emanuel BS, Gerstein MB, Weissman SM, Snyder M (21 March 2006). "High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays". Proc Natl Acad Sci U S A 103 (12): 4534–4539. doi:10.1073/pnas.0511340103. PMC 1450206. PMID 16537408.
- ↑ 18.0 18.1 18.2 18.3 18.4 Bejjani BA, Shaffer LG (2006) Applications of array-based comparative genomic hybridization to clinical diagnostics. J Mol Diagn 8:528-533.
- ↑ Shinawi M, Cheung SW (2008) The array CGH and its clinical applications. Drug Discovery Today 13:760-769.
- ↑ Fiegler H, Carr P, Douglas EJ, Burford DC, Hunt S, Scott CE, Smith J, Vetrie D, Gorman P, Tomlinson IP, Carter NP (2003) DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes Chromosomes Cancer 36:361-374.
- ↑ Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF, Jeffrey SS, Botstein D, Brown PO (1999) Genome-wide analysis of DNA copy number changes using cDNA microarrays. Nat Genet 23:41-46.
- ↑ Carvalho B, Ouwerkerk E, Meijer GA, Ylstra B. (2004) High resolution microarray comparative genomic hybridization analysis using spotted oligonucleotides. J Clin Pathol 57: 644–646.
- ↑ Weiss MM, Kuipers EJ, Meuwissen SG, van Diest PJ, Meijer GA (2003) Comparative genomic hybridization as a supportive tool in diagnostic pathology. J Clin Pathol 56:522-527.
- ↑ 24.0 24.1 24.2 Forozan F, Karhu R, Kononen J, Kallioniemi A, Kallioniemi OP (1997) Genome screening by comparative genomic hybridization. Trends Genet 13:405-409.
- ↑ Baudis M & Cleary ML (2001). Progenetix.net: an online repository for molecular cytogenetic aberration data. Bioinformatics, 17(12), 1228–1229.
- ↑ Cai H, Kumar N, Ai N, Gupta S, Rath P & Baudis M. (2014). Progenetix: 12 years of oncogenomic data curation. Nucleic Acids Research, 42(D1), D1055–D1062.
- ↑ 27.0 27.1 Levy B, Dunn TM, Kern JH, Hirschhorn K, Kardon NB (2002) Delineation of the dup5q phenotype by molecular cytogenetic analysis in a patient with dup5q/del 5p (Cri du Chat). Am J Med Genet 108:192-197.
- ↑ Mraz, M.; Stano Kozubik, K.; Plevova, K.; Musilova, K.; Tichy, B.; Borsky, M.; Kuglik, P.; Doubek, M.; Brychtova, Y.; Mayer, J.; Pospisilova, S. (2013). "The origin of deletion 22q11 in chronic lymphocytic leukemia is related to the rearrangement of immunoglobulin lambda light chain locus". Leukemia Research 37 (7): 802–808. doi:10.1016/j.leukres.2013.03.018. PMID 23608880.
- ↑ Cai H, Kumar N, & Baudis M. (2012). arrayMap: a reference resource for genomic copy number imbalances in human malignancies. PLoS ONE, 7(5), e36944. doi:10.1371/journal.pone.0036944
- ↑ L’Hermine AC, Aboura A, Brisset S, Cuisset L, Castaigne V, Labrune P, Frydman R, Tachdjian G. (2003) Fetal phenotype of Prader–Willi syndrome due to maternal disomy for chromosome 15. Prenat Diagn 23:938-943.
- ↑ Vissers LE, de Vries BB, Osoegawa K, Janssen IM, Feuth T, Choy CO, Straatman H, van der Vliet W, Huys EH, van Rijk A, Smeets D, van Ravenswaaij-Arts CM, Knoers NV, van der Burgt I, de Jong PJ, Brunner HG, Geurts van Kessel A, Schoenmakers EF, Veltman JA (2003) Array-based comparative genomic hybridization for the genome-wide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet 73:1261-1270.
- ↑ Fiorentino F (2012) Array comparative genomic hybridization: its role in preimplantation genetic diagnosis. Current Opinion in Obstetrics and Gynecology 24:203-209.
- ↑ Lee CN, Lin SY, Lin CH, Shih JC, Lin TH, Su YN (2012) Clinical utility of array comparative genomic hybridization for prenatal diagnosis: a cohort study of 3171 pregnancies. BJOG: An International Journal of Obstetrics & Gynaecology 119:614-625.
- ↑ Weiss MM, Hermsen MAJA, Meijer GA, van Grieken NCT, Baak JPA, Kuipers EJ, van Deist PJ (1999) Comparative genomic hybridization. J Clin Pathol: Mol Pathol 52:243-251.
- ↑ du Manoir S, Schrock E, Bentz M, Speicher MR, Joos S, Ried T, Lichter P, Cremer T (1995) Quantitative analysis of comparative genomic hybridization. Cytometry 19:27-41.
- ↑ Shaw CJ, Stankiewicz P, Bien-Willner G, Bello SC, Shaw CA, Carrera M, Perez Jurado L, Estivill X, Lupski JR. (2004) Small marker chromosomes in two patients with segmental aneusomy for proximal 17p. Hum Genet 115:1-7.
External links
- Virtual Grand Rounds: "Differentiating Microarray Technologies and Related Clinical Implications" by Arthur Beaudet, MD
- Progenetix database: A large, continuously updated collection of CGH and aCGH and associated clinical data (ca. 32'000 cases, September 2014).
- CGH article tracker: An attempt to list all (a)CGH publications containing original case data (2538 as of September 2014). This is part of the Progenetix project.
- arrayMap repository: Continuously expanded collection cancer genome array datasets, with per-array and aggregated data visualisation (ca. 64'000 arrays, September 2014).
- NCBI's Cancer Chromosomes: Cancer Chromosome is an integral part of NCBI's Entrez system, which combines several databases with (molecular-) cytogenetic data.