Chemical thermodynamics
Chemical thermodynamics is the study of the interrelation of heat and work with chemical reactions or with physical changes of state within the confines of the laws of thermodynamics. Chemical thermodynamics involves not only laboratory measurements of various thermodynamic properties, but also the application of mathematical methods to the study of chemical questions and the spontaneity of processes.
The structure of chemical thermodynamics is based on the first two laws of thermodynamics. Starting from the first and second laws of thermodynamics, four equations called the "fundamental equations of Gibbs" can be derived. From these four, a multitude of equations, relating the thermodynamic properties of the thermodynamic system can be derived using relatively simple mathematics. This outlines the mathematical framework of chemical thermodynamics.[1]
History

In 1865, the German physicist Rudolf Clausius, in his Mechanical Theory of Heat, suggested that the principles of thermochemistry, e.g. the heat evolved in combustion reactions, could be applied to the principles of thermodynamics.[2] Building on the work of Clausius, between the years 1873-76 the American mathematical physicist Willard Gibbs published a series of three papers, the most famous one being the paper On the Equilibrium of Heterogeneous Substances. In these papers, Gibbs showed how the first two laws of thermodynamics could be measured graphically and mathematically to determine both the thermodynamic equilibrium of chemical reactions as well as their tendencies to occur or proceed. Gibbs’ collection of papers provided the first unified body of thermodynamic theorems from the principles developed by others, such as Clausius and Sadi Carnot.
During the early 20th century, two major publications successfully applied the principles developed by Gibbs to chemical processes, and thus established the foundation of the science of chemical thermodynamics. The first was the 1923 textbook Thermodynamics and the Free Energy of Chemical Substances by Gilbert N. Lewis and Merle Randall. This book was responsible for supplanting the chemical affinity for the term free energy in the English-speaking world. The second was the 1933 book Modern Thermodynamics by the methods of Willard Gibbs written by E. A. Guggenheim. In this manner, Lewis, Randall, and Guggenheim are considered as the founders of modern chemical thermodynamics because of the major contribution of these two books in unifying the application of thermodynamics to chemistry.[1]
Overview
The primary objective of chemical thermodynamics is the establishment of a criterion for the determination of the feasibility or spontaneity of a given transformation.[3] In this manner, chemical thermodynamics is typically used to predict the energy exchanges that occur in the following processes:
- Chemical reactions
- Phase changes
- The formation of solutions
The following state functions are of primary concern in chemical thermodynamics:
- Internal energy (U)
- Enthalpy (H)
- Entropy (S)
- Gibbs free energy (G)
Most identities in chemical thermodynamics arise from application of the first and second laws of thermodynamics, particularly the law of conservation of energy, to these state functions.
The 3 laws of thermodynamics:
- The energy of the universe is constant.
- In any spontaneous process, there is always an increase in entropy of the universe
- The entropy of a perfect crystal at 0 Kelvin is zero
Chemical energy
Chemical energy is the potential of a chemical substance to undergo a transformation through a chemical reaction or to transform other chemical substances. Breaking or making of chemical bonds involves energy, which may be either absorbed or evolved from a chemical system.
Energy that can be released (or absorbed) because of a reaction between a set of chemical substances is equal to the difference between the energy content of the products and the reactants. This change in energy is called the change in internal energy of a chemical reaction. Where 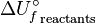 is the internal energy of formation of the reactant molecules that can be calculated from the bond energies of the various chemical bonds of the molecules under consideration and
is the internal energy of formation of the reactant molecules that can be calculated from the bond energies of the various chemical bonds of the molecules under consideration and 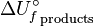 is the internal energy of formation of the product molecules. The internal energy change of a process is equal to the heat change if it is measured under conditions of constant volume, as in a closed rigid container such as a bomb calorimeter. However, under conditions of constant pressure, as in reactions in vessels open to the atmosphere, the measured heat change is not always equal to the internal energy change, because pressure-volume work also releases or absorbs energy. (The heat change at constant pressure is called the enthalpy change; in this case the enthalpy of formation).
is the internal energy of formation of the product molecules. The internal energy change of a process is equal to the heat change if it is measured under conditions of constant volume, as in a closed rigid container such as a bomb calorimeter. However, under conditions of constant pressure, as in reactions in vessels open to the atmosphere, the measured heat change is not always equal to the internal energy change, because pressure-volume work also releases or absorbs energy. (The heat change at constant pressure is called the enthalpy change; in this case the enthalpy of formation).
Another useful term is the heat of combustion, which is the energy released due to a combustion reaction and often applied in the study of fuels. Food is similar to hydrocarbon fuel and carbohydrate fuels, and when it is oxidized, its caloric content is similar (though not assessed in the same way as a hydrocarbon fuel — see food energy).
In chemical thermodynamics the term used for the chemical potential energy is chemical potential, and for chemical transformation an equation most often used is the Gibbs-Duhem equation.
Chemical reactions
In most cases of interest in chemical thermodynamics there are internal degrees of freedom and processes, such as chemical reactions and phase transitions, which always create entropy unless they are at equilibrium, or are maintained at a "running equilibrium" through "quasi-static" changes by being coupled to constraining devices, such as pistons or electrodes, to deliver and receive external work. Even for homogeneous "bulk" materials, the free energy functions depend on the composition, as do all the extensive thermodynamic potentials, including the internal energy. If the quantities { Ni }, the number of chemical species, are omitted from the formulae, it is impossible to describe compositional changes.
Gibbs function
For a "bulk" (unstructured) system they are the last remaining extensive variables. For an unstructured, homogeneous "bulk" system, there are still various extensive compositional variables { Ni } that G depends on, which specify the composition, the amounts of each chemical substance, expressed as the numbers of molecules present or (dividing by Avogadro's number), the numbers of moles
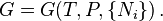
For the case where only PV work is possible
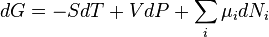
in which μi is the chemical potential for the i-th component in the system
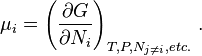
The expression for dG is especially useful at constant T and P, conditions which are easy to achieve experimentally and which approximates the condition in living creatures
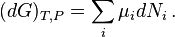
Chemical affinity
While this formulation is mathematically defensible, it is not particularly transparent since one does not simply add or remove molecules from a system. There is always a process involved in changing the composition; e.g., a chemical reaction (or many), or movement of molecules from one phase (liquid) to another (gas or solid). We should find a notation which does not seem to imply that the amounts of the components ( Ni } can be changed independently. All real processes obey conservation of mass, and in addition, conservation of the numbers of atoms of each kind. Whatever molecules are transferred to or from should be considered part of the "system".
Consequently we introduce an explicit variable to represent the degree of advancement of a process, a progress variable ξ for the extent of reaction (Prigogine & Defay, p. 18; Prigogine, pp. 4–7; Guggenheim, p. 37.62), and to the use of the partial derivative ∂G/∂ξ (in place of the widely used "ΔG", since the quantity at issue is not a finite change). The result is an understandable expression for the dependence of dG on chemical reactions (or other processes). If there is just one reaction
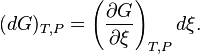
If we introduce the stoichiometric coefficient for the i-th component in the reaction
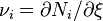
which tells how many molecules of i are produced or consumed, we obtain an algebraic expression for the partial derivative
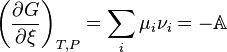
where, (De Donder; Progoine & Defay, p. 69; Guggenheim, pp. 37,240), we introduce a concise and historical name for this quantity, the "affinity", symbolized by A, as introduced by Théophile de Donder in 1923. The minus sign comes from the fact the affinity was defined to represent the rule that spontaneous changes will ensue only when the change in the Gibbs free energy of the process is negative, meaning that the chemical species have a positive affinity for each other. The differential for G takes on a simple form which displays its dependence on compositional change
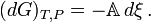
If there are a number of chemical reactions going on simultaneously, as is usually the case
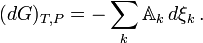
a set of reaction coordinates { ξj }, avoiding the notion that the amounts of the components ( Ni } can be changed independently. The expressions above are equal to zero at thermodynamic equilibrium, while in the general case for real systems, they are negative because all chemical reactions proceeding at a finite rate produce entropy. This can be made even more explicit by introducing the reaction rates dξj/dt. For each and every physically independent process (Prigogine & Defay, p. 38; Prigogine, p. 24)
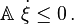
This is a remarkable result since the chemical potentials are intensive system variables, depending only on the local molecular milieu. They cannot "know" whether the temperature and pressure (or any other system variables) are going to be held constant over time. It is a purely local criterion and must hold regardless of any such constraints. Of course, it could have been obtained by taking partial derivatives of any of the other fundamental state functions, but nonetheless is a general criterion for (−T times) the entropy production from that spontaneous process; or at least any part of it that is not captured as external work. (See Constraints below.)
We now relax the requirement of a homogeneous “bulk” system by letting the chemical potentials and the affinity apply to any locality in which a chemical reaction (or any other process) is occurring. By accounting for the entropy production due to irreversible processes, the inequality for dG is now replaced by an equality
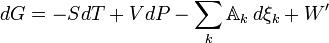
or
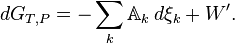
Any decrease in the Gibbs function of a system is the upper limit for any isothermal, isobaric work that can be captured in the surroundings, or it may simply be dissipated, appearing as T times a corresponding increase in the entropy of the system and/or its surrounding. Or it may go partly toward doing external work and partly toward creating entropy. The important point is that the extent of reaction for a chemical reaction may be coupled to the displacement of some external mechanical or electrical quantity in such a way that one can advance only if the other one also does. The coupling may occasionally be rigid, but it is often flexible and variable.
Solutions
In solution chemistry and biochemistry, the Gibbs free energy decrease (∂G/∂ξ, in molar units, denoted cryptically by ΔG) is commonly used as a surrogate for (−T times) the entropy produced by spontaneous chemical reactions in situations where there is no work being done; or at least no "useful" work; i.e., other than perhaps some ± PdV. The assertion that all spontaneous reactions have a negative ΔG is merely a restatement of the fundamental thermodynamic relation, giving it the physical dimensions of energy and somewhat obscuring its significance in terms of entropy. When there is no useful work being done, it would be less misleading to use the Legendre transforms of the entropy appropriate for constant T, or for constant T and P, the Massieu functions −F/T and −G/T respectively.
Non equilibrium
Generally the systems treated with the conventional chemical thermodynamics are either at equilibrium or near equilibrium. Ilya Prigogine developed the thermodynamic treatment of open systems that are far from equilibrium. In doing so he has discovered phenomena and structures of completely new and completely unexpected types. His generalized, nonlinear and irreversible thermodynamics has found surprising applications in a wide variety of fields.
The non equilibrium thermodynamics has been applied for explaining how ordered structures e.g. the biological systems, can develop from disorder. Even if Onsager's relations are utilized, the classical principles of equilibrium in thermodynamics still show that linear systems close to equilibrium always develop into states of disorder which are stable to perturbations and cannot explain the occurrence of ordered structures.
Prigogine called these systems dissipative systems, because they are formed and maintained by the dissipative processes which take place because of the exchange of energy between the system and its environment and because they disappear if that exchange ceases. They may be said to live in symbiosis with their environment.
The method which Prigogine used to study the stability of the dissipative structures to perturbations is of very great general interest. It makes it possible to study the most varied problems, such as city traffic problems, the stability of insect communities, the development of ordered biological structures and the growth of cancer cells to mention but a few examples.
System constraints
In this regard, it is crucial to understand the role of walls and other constraints, and the distinction between independent processes and coupling. Contrary to the clear implications of many reference sources, the previous analysis is not restricted to homogeneous, isotropic bulk systems which can deliver only PdV work to the outside world, but applies even to the most structured systems. There are complex systems with many chemical "reactions" going on at the same time, some of which are really only parts of the same, overall process. An independent process is one that could proceed even if all others were unaccountably stopped in their tracks. Understanding this is perhaps a “thought experiment” in chemical kinetics, but actual examples exist.
A gas reaction which results in an increase in the number of molecules will lead to an increase in volume at constant external pressure. If it occurs inside a cylinder closed with a piston, the equilibrated reaction can proceed only by doing work against an external force on the piston. The extent variable for the reaction can increase only if the piston moves, and conversely, if the piston is pushed inward, the reaction is driven backwards.
Similarly, a redox reaction might occur in an electrochemical cell with the passage of current in wires connecting the electrodes. The half-cell reactions at the electrodes are constrained if no current is allowed to flow. The current might be dissipated as joule heating, or it might in turn run an electrical device like a motor doing mechanical work. An automobile lead-acid battery can be recharged, driving the chemical reaction backwards. In this case as well, the reaction is not an independent process. Some, perhaps most, of the Gibbs free energy of reaction may be delivered as external work.
The hydrolysis of ATP to ADP and phosphate can drive the force times distance work delivered by living muscles, and synthesis of ATP is in turn driven by a redox chain in mitochondria and chloroplasts, which involves the transport of ions across the membranes of these cellular organelles. The coupling of processes here, and in the previous examples, is often not complete. Gas can leak slowly past a piston, just as it can slowly leak out of a rubber balloon. Some reaction may occur in a battery even if no external current is flowing. There is usually a coupling coefficient, which may depend on relative rates, which determines what percentage of the driving free energy is turned into external work, or captured as "chemical work"; a misnomer for the free energy of another chemical process.
See also
References
- ↑ 1.0 1.1 Ott, Bevan J.; Boerio-Goates, Juliana (2000). Chemical Thermodynamics – Principles and Applications. Academic Press. ISBN 0-12-530990-2.
- ↑ Clausius, R. (1865). The Mechanical Theory of Heat – with its Applications to the Steam Engine and to Physical Properties of Bodies. London: John van Voorst, 1 Paternoster Row. MDCCCLXVII.
- ↑ Klotz, I. (1950). Chemical Thermodynamics. New York: Prentice-Hall, Inc.
Further reading
- Herbert B. Callen (1960). Thermodynamics. Wiley & Sons. The clearest account of the logical foundations of the subject. ISBN 0-471-13035-4. Library of Congress Catalog No. 60-5597
- Ilya Prigogine & R. Defay, translated by D.H. Everett; Chapter IV (1954). Chemical Thermodynamics. Longmans, Green & Co. Exceptionally clear on the logical foundations as applied to chemistry; includes non-equilibrium thermodynamics.
- Ilya Prigogine (1967). Thermodynamics of Irreversible Processes, 3rd ed. Interscience: John Wiley & Sons. A simple, concise monograph explaining all the basic ideas. Library of Congress Catalog No. 67-29540
- E.A. Guggenheim (1967). Thermodynamics: An Advanced Treatment for Chemists and Physicists, 5th ed. North Holland; John Wiley & Sons (Interscience). A remarkably astute treatise. Library of Congress Catalog No. 67-20003
- Th. De Donder (1922). Bull. Ac. Roy. Belg. (Cl. Sc.) (5) 7: 197, 205. Missing or empty
|title=(help)
External links
- Chemical Thermodynamics - University of North Carolina
- Chemical energetics (Introduction to thermodynamics and the First Law)
- Thermodynamics of chemical equilibrium (Entropy, Second Law and free energy)
| |||||||||||||||||||||||||||||||||
