Bleach and recycle
The "bleach and recycle" process is used within the retina to ensure that the chromophore, 11-cis retinal, is present within opsin molecules in sufficient quantities to allow phototransduction to occur. It uses vitamin A (retinol) derivatives.
Retinoid transport to the retina
Vitamin A must be consumed in the diet, as it is not synthesisable by the body. It may, however, be consumed indirectly, in the form of carotenoids such as beta carotene, which can be cleaved to form two retinol molecules. It is absorbed in the gut and is transported around the body via two pathways. In the first, and most predominant, it is esterified with a fatty acid to form a retinyl ester, and packaged into a chylomicron. In the second minor pathway, it is bound to Retinol Binding Protein (RBP) and Transthyretin, which prevents its filtration in the glomeruli. It is via this RBP-Transthyretin pathway that the retina receives most of its retinoids.
As in transport via the RBP-Transthyretin pathway, retinoids must always be bound to chaperone molecules, for several reasons. Retinoids are toxic, insoluble in aqueous solutions, and prone to oxidation, and as such they must be bound and protected when within the body. The body uses a variety of chaperones, particularly in the retina, to transport retinoids.
Bleach and recycle pathway
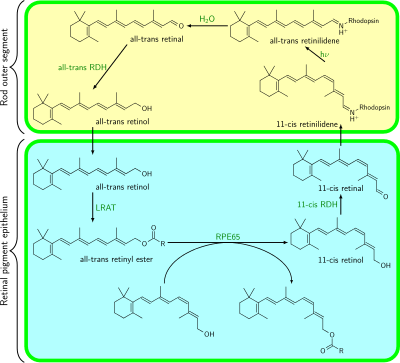
When a photon of light is absorbed, 11-cis retinal is transformed to all-trans retinal, and it moves to the exit site of rhodopsin. It will not leave the opsin protein until another fresh chromophore comes to replace it, except for in the ABCR pathway. Whilst still bound to the opsin, all-trans retinal is transformed into all-trans retinol by all-trans Retinol Dehydrogenase. It then proceeds to the cell membrane of the rod, where it is chaperoned to the Retinal Pigment Epithelium (RPE) by Interphotoreceptor Retinoid Binding Protein (IRBP). It then enters the RPE cells, and is transferred to the Cellular Retinol Binding Protein (CRBP) chaperone.
When inside the RPE cell, bound to CRBP, the all-trans retinol is esterified by Lecithin Retinol Acyltransferase (LRAT) to form a retinyl ester. The retinyl esters of the RPE are chaperoned by a protein known as RPE65. It is in this form that the RPE stores most of its retinoids, as the RPE stores 2-3 times more retinoids than the neural retina itself. When further chromophore is required, the retinyl esters are acted on by isomerohydrolase to produce 11-cis retinol, which is transferred to the Cellular Retinaldehyde Binding Protein (CRALBP). 11-cis retinol is transformed into 11-cis retinal by 11-cis Retinol Dehydrogenase, then it is shipped back to the rod photoreceptors via IRBP. There, it replaces the spent chromophore in opsin molecules, allowing the opsin to function again as a light receptor.
ABCR pathway
Under some circumstances, the spent chromophore may leave the opsin molecule prior to its replacement, when it is bound to the ABCA4 protein (also known as ABCR), when it is also transformed to all-trans retinol, and then leaves the photoreceptor outer segment via the IRBP chaperone. It then follows the conventional pathway of recycling. It is from this pathway that the presence of opsin without a chromophore can be explained.
RGR regulation of recycling
The recycling process can be regulated by the Retinol G-protein Receptor system. When light is incident upon the Retinol G-protein Receptor (RGR) the recycling of chromophore in the RPE is accelerated. This mechanism provides additional chromophore after intense bleaches, and can be seen as an important mechanism in the early phases of dark adaptation and chromophore replenishment.
Alternate cone pathway
It is believed that as well as this pathway of chromophore recycling described above, that cone photoreceptors have an alternate pathway of chromophore recycling which uses Muller's cells within the retina to hasten chromophore recycling. In this pathway, cones reduce all-trans retinal to all-trans retinol via all-trans Retinol Dehydrogenase, then transport all-trans retinol to Muller's cells. There, it is transformed into 11-cis retinol by all-trans retinol isomerase, and can either be stored as retinyl esters within Muller's cells, or transported back to the cone photoreceptors, where it is transformed from 11-cis retinol to 11-cis retinal by 11-cis Retinal Dehydrogenase. This pathway helps explain the rapid dark adaptation in the cone system, and the presence of 11-cis Retinal Dehydrogenase in cone photoreceptors, as it is not found in rods, only in the RPE.
Leber's congenital amaurosis
A possible mechanism for Leber's congenital amaurosis has been proposed as the deficiency of RPE65. If one was lacking in RPE65 the RPE would be unable to store retinyl esters, and thus the bleach and recycle pathway would be compromised. This would lead predominantly to night blindness (as the rods have only one mechanism, via the RPE, to regenerate their chromophore) in the early stages of the disease, at which stage the cone photoreceptors would be spared, as they have the alternate Muller cell pathway for photopigment regeneration.
In the later stages of the disease, general retinopathy is observed as the rod photoreceptors lose their ability to signal the presence of light, because of their deficiency of 11-cis retinal chromophore. The rods will thus continually secrete glutamate neurotransmitter, and after a time the Muller cells will be unable to mop up the excess glutamate. The glutamate levels will build up within the retina, where they will reach neurotoxic levels and the retina will begin to be destroyed.
The RPE65 deficiency would be genetic in origin, and is only one of many proposed possible pathophysiologies of the disease.