Birt–Hogg–Dubé syndrome
| Birt–Hogg–Dubé syndrome | |
|---|---|
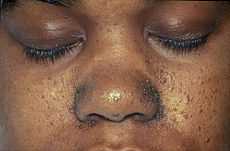
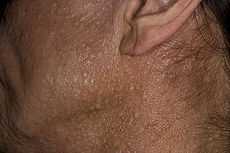 The characteristic fibrofolliculomas of Birt-Hogg-Dubé syndrome seen on a person's face. | |
| Classification and external resources | |
| OMIM | 135150 |
| DiseasesDB | 33274 |
| eMedicine | derm/622 |
| GeneReviews | |
Birt–Hogg–Dubé syndrome (BHD), also Hornstein–Birt–Hogg–Dubé syndrome, Hornstein–Knickenberg syndrome, and fibrofolliculomas with trichodiscomas and acrochordons[1] is a human autosomal dominant genetic disorder that can cause susceptibility to kidney cancer, renal and pulmonary cysts, and noncancerous tumors of the hair follicles, called fibrofolliculomas. The symptoms seen in each family are unique, and can include any combination of the three symptoms. Fibrofolliculomas are the most common manifestation, found on the face and upper trunk in over 80% of people with BHD over the age of 40. Pulmonary cysts are equally common (84%), but only 24% of people with BHD eventually experience a collapsed lung (spontaneous pneumothorax). Kidney tumors, both cancerous and benign, occur in 14–34% of people with BHD; the associated kidney cancers are often rare hybrid tumors.
Any of these conditions that occurs in a family can indicate a diagnosis of Birt–Hogg–Dubé syndrome, though it is only confirmed by a genetic test for a mutation in the FLCN gene, which codes for the protein folliculin. Though its function is not fully understood, it appears to be a tumor suppressor gene that restricts cell growth and division. Versions of FLCN have been found in other animals, including fruit flies, German shepherds, rats, and mice. The disease was discovered in 1977, but the connection with FLCN was not elucidated until 2002, after kidney cancer, collapsed lungs, and pulmonary cysts were all definitively connected to BHD.
Birt–Hogg–Dubé syndrome can manifest similarly to other diseases, which must be ruled out when making a diagnosis. These include tuberous sclerosis, which causes skin lesions similar to fibrofolliculomas, and Von Hippel-Lindau disease, which causes hereditary kidney cancers. Once diagnosed, people with BHD are treated preventatively, with monitoring of kidneys and lungs using medical imaging. Fibrofolliculomas can be removed surgically and pneumothorax and kidney cancer are treated according to the normal standard of care.
Signs and symptoms
Skin

Birt–Hogg–Dubé syndrome affects the skin and increases the risk of tumors in the kidneys and lungs. The condition is characterized by multiple noncancerous dome-shaped tumors of the hair follicles (fibrofolliculomas), particularly on the face, neck, and—more rarely—the upper chest.[2] The fibrofolliculomas are generally described as having an opaque white color[3] or a yellowish tone[4] and have a waxy, smooth texture.[3] The tumors are always found on and around the nose and on and behind the outer ear. Typically, they first appear in a person's 20s or 30s, and are found in more than 80% of people with the syndrome above the age of 40.[2] The tumors become larger and more numerous over time.[5] Tumors differ between individuals: they may appear merged in plaques, look similar to a comedo with a plug of keratin, or include epidermoid cysts. A large number of tumors on the face can be associated with hyperseborrhea (abnormally elevated sebum production).[2] The presence of fibrofolliculomas on a person's face can cause significant psychological distress.[6]
Other tumors can include trichodiscomas (tumors of the hair disc, which may be identical to fibrofolliculomas), angiofibromas, and perifollicular fibromas.[5] However, angiofibromas are more common in tuberous sclerosis.[6] Along with the tumors, other skin conditions are seen in people with Birt–Hogg–Dubé syndrome. Approximately 40% of people or families with the disease have papules in their mouth, which can be located on the cheeks (buccal mucosa), tongue, gums, or lips. Either white or mucosa-colored, they are discrete, small, and soft and consist of fibrous tissue covered in thickened epithelium.[2] Collagenomas of the skin are also found in some families.[5] Many people with BHD have skin lesions that appear to be acrochordons (skin tags), but may instead be fibrofolliculomas. These lesions are usually found in the armpit, on the eyelids, and in folds of skin.[3] Not all individuals develop the facial tumors; some families with the mutation that causes BHD develop only kidney tumors or spontaneous pneumothorax.[5]
Kidneys
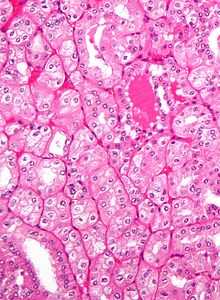
People over 20 years of age with Birt–Hogg–Dubé syndrome have an increased risk of developing slow-growing kidney tumors (chromophobe renal carcinoma and renal oncocytoma, respectively), kidney cysts, and possibly tumors in other organs and tissues.[2] These tumors often occur in both kidneys and in multiple locations in each kidney.[5] The average number of kidney tumors found in a person with BHD is 5.3, though up to 28 tumors have been found.[7] Hybrid oncocytoma/chromophobe carcinoma, found in 50% of cases,[8] is the most commonly found cancer, followed by chromophobe renal carcinoma, clear cell renal carcinoma, renal oncocytoma, and papillary renal cell carcinoma.[5][8] People over 40 years old and men are more likely to develop kidney tumors, which are diagnosed at a median age of 48.[2][5] Kidney cancer associated with BHD have been diagnosed in people at ages as young as 20.[6]
In general, people with Birt–Hogg–Dubé syndrome are at roughly seven times the risk of kidney cancer compared to the unaffected population. Estimates of the incidence among people with the disease range from 14%–34%.[2][8] Rarely, it is associated with clear cell renal cell carcinoma and papillary renal cell carcinoma.[5] If it develops in someone with BHD, renal cell carcinoma occurs later in life and has a poor prognosis.[9] Though the types of tumor typically associated with BHD are considered less aggressive, cases of advanced or metastatic kidney cancer have been observed in people with the syndrome. Both benign and cancerous tumors can reduce kidney function over time as they grow larger.[8]
Lungs
Along with fibrofolliculomas and kidney tumors, affected individuals frequently develop cysts (blebs or bullae) in the subpleural lung base or intraparenchymal space that may rupture and cause an abnormal collection of air in the chest cavity (pneumothorax), which could result in the collapse of a lung.[5][10] The cysts do not cause other symptoms[5] and lung function is usually normal.[6] More than 83% of people with Birt–Hogg–Dubé have cysts, however, the syndrome does not cause conditions like progressive chronic obstructive pulmonary disease (COPD) or generalized respiratory failure,[2] though it does cause emphysema.[4] Spontaneous, sometimes recurrent,[5] pneumothorax occurs far more often and at a younger age with Birt–Hogg–Dubé than in the unaffected population. Approximately 24% of people with the disease suffer at least one spontaneous pneumothorax, 30 times the occurrence in unaffected people. Though pneumothorax caused by Birt–Hogg–Dubé often occurs in middle age, at a median age of 38, 17% of affected people have a spontaneous pneumothorax before turning 40.[2] Pneumothoraces have been seen in people as young as 7 and 16 years of age.[6] Some families have a form of BHD that only affects the lungs.[11]
Other organs
Thyroid nodules[2] have been associated with the Birt–Hogg–Dubé phenotype, present in 65% of individuals and 90% of families with the syndrome.[2] However, a connection between BHD and thyroid cancer has not been substantiated.[3] Other conditions have been reported to be associated but may not be caused by the mutation in FLCN or may not be related at all. These include multinodular goiter, medullary thyroid carcinoma, parotid oncocytoma, colonic polyposis,[12] connective tissue nevus, lipomas, angiolipomas, parathyroid adenomas, flecked chorioretinopathy, neurothekeoma, meningiomas, angiofibromas of the face,[3] trichoblastomas, cutaneous focal mucinosis, cutaneous leiomyoma, breast cancer, tonsillar cancer, colorectal cancer, sarcoma of the leg, lung cancer, melanoma, dermatofibrosarcoma protuberans, basal cell carcinoma, cutaneous leiomyosarcoma, and squamous cell carcinoma.[6]
Pathophysiology
Genetics

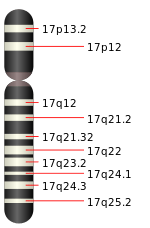
An association with the folliculin (FLCN) gene was first reported in 2002.[13] This 14-exon gene is located on the short arm of chromosome 17 (17p11.2) and has a cytosine-rich region in exon 11 particularly susceptible to mutation.[2][5] The most common mutation in this region is the insertion or deletion of a cytosine residue, found in 53% of Birt–Hogg–Dubé families. There is no significant difference in the symptoms experienced by families with an insertion at that location compared to those who have a deletion. However, mutations in FLCN associated with Birt–Hogg–Dubé syndrome are heterogeneous, and are often nonsense mutations or frameshift mutations that cause early truncation of the protein product at the carboxy-terminus.[14][3] Very rarely, missense mutations are observed.[6] Mutations are often passed from one generation to the next in an autosomal dominant fashion but can occur as a new mutation in an individual with no prior family history (a de novo mutation).[15] The children of an affected parent each have a 50% chance of having the disease. Birt–Hogg–Dubé syndrome has very high penetrance.[5] A correlation between different FLCN genotypes and phenotypes has not been discovered.[15]
Function
FLCN creates a protein called folliculin that has two isoforms.[3] It appears to act as a tumor suppressor and is expressed strongly in the skin, distal nephrons, and type I pneumocytes.[5] It has also been found in the parotid gland, brain, breast, pancreas, prostate, and ovaries.[8][4] Tumor suppressors normally prevent cells from growing and dividing too rapidly or in an uncontrolled way. Mutations in the FLCN gene may interfere with the ability of folliculin to restrain cell growth and division, leading to the formation of noncancerous and cancerous tumors. Recent studies suggest that folliculin accomplishes this function through its involvement with cellular metabolism, possibly through modulation of the mTOR (mammalian target of rapamycin) pathway and/or oxidative phosphorylation in mitochondria.[16] Folliculin interacts with FNIP1 and FNIP2 (FLCN-interacting protein) to form a complex with AMP-activated protein kinase.[8][4] Folliculin's participation in the mTOR pathway may explain the similarity in phenotype between BHD syndrome, Cowden syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome.[3]
Most of the cancer-causing mutations cause the protein to be truncated at the carboxy-terminus.[8] The C-terminal end of folliculin has shown to be the domain through which it interacts with FNIP1, and thereby possibly the mTOR pathway.[14] FLCN is highly conserved in vertebrates—it is very similar between many vertebrate species.[5] The 508th amino acid, normally lysine, is affected by a missense mutation in some people with Birt–Hogg–Dubé syndrome. The lysine at this position is found to be conserved between invertebrate and vertebrate orthologs of folliculin, indicating that it is important to the protein's function.[14]
People with Birt–Hogg–Dubé syndrome are born with one mutated copy of the FLCN gene in each cell.[6] Haploinsufficiency—only having one functional copy of the FLCN gene—is enough to cause the fibrofolliculomas and pulmonary cysts, however, one copy of the gene is enough to keep kidney cells in check.[1] During their lifetime, random mutations might inactivate the normal copy of the gene in a subset of cells. When this occurs, the result is that these cells have no functional copies of the FLCN gene, allowing the cells grow out of control. This loss of heterozygosity is a common mechanism in cancer, and it is frequently detected in the renal cancers associated with BHD. The molecular genetic defects in renal tumors of people with BHD are different from two other similar kidney tumors, chromophobe renal cell carcinoma and renal oncocytoma. BHD-associated tumorigenesis differs between the kidney, where loss of FLCN heterozygosity is responsible for cancers, and the skin, where FLCN is strongly expressed in heterozygotes.[6] FLCN has been found to be overexpressed in fibrofolliculoma tissue and to have very low levels of expression in affected kidneys.[4] Furthermore, the mTOR pathway is shown to be activated in tumor tissue from both humans and mice.[7]
Diagnosis
BHD can be suggested by clinical findings but is definitively diagnosed by molecular genetic testing to detect mutations in the FLCN gene. The classical clinical triad includes benign growths of the hair follicles; pulmonary cysts and spontaneous pneumothorax; and bilateral, multifocal renal tumors.[5]
Clinical triad
The cutaneous manifestations of Birt–Hogg–Dubé were originally described as fibrofolliculomas (abnormal growths of a hair follicle), trichodiscomas (hamartomatous lesions with a hair follicle at the periphery, often found on the face), and acrochordons (skin tags). Cutaneous manifestations are confirmed by histology. Most individuals (89%) with BHD are found to have multiple cysts in both lungs, and 24% have had one or more episodes of pneumothorax. The cysts can be detected by chest CT scan. Renal tumors can manifest as multiple types of renal cell carcinoma, but certain pathological subtypes (including chromophobe, oncocytoma, and oncocytic hybrid tumors) are more commonly seen. Although the original syndrome was discovered on the basis of cutaneous findings, it is now recognized that individuals with Birt–Hogg–Dubé may only manifest the pulmonary and/or renal findings, without any skin lesions. Though these signs indicate BHD, it is only confirmed with a genetic test for FLCN mutations.[5]
Genetic testing
FLCN mutations are detected by sequencing in 88% of probands with Birt–Hogg–Dubé syndrome. This means that some people with the clinical diagnosis have mutations that are not detectable by current technology, or that mutations in another currently unknown gene could be responsible for a minority of cases. In addition, amplifications and deletions in exonic regions are also tested. Genetic testing can be useful to confirm the clinical diagnosis of and to provide a means of determining other at-risk individuals in a family even if they have not yet developed BHD symptoms.[5][6]
Differential diagnosis
Birt–Hogg–Dubé can be difficult to diagnose from symptoms alone, because hereditary renal cancers, pneumothorax, and cutaneous tumors occur with other syndromes. Hereditary bilateral, multifocal kidney tumors similar to those seen in BHDcan occur with von Hippel–Lindau disease (clear cell renal cell carcinoma), hereditary papillary renal cancer (papillary renal cell carcinoma), and hereditary leiomyomatosis and renal cell cancer syndrome. They are differentiated with examination of the tumors' histology.[5]
Hereditary recurrent pneumothorax or pulmonary cysts are associated with Marfan syndrome, Ehlers–Danlos syndrome, Tuberous Sclerosis Complex (TSC), alpha1-antitrypsin deficiency, and cystic fibrosis. Non-hereditary recurrent pneumothorax and/or pulmonary cysts can occur with Langerhans cell histiocytosis and lymphangioleiomyomatosis. These conditions are differentiated from Birt–Hogg–Dubé through examining the patient history and performing a physical examination.[5] In women suspected to have the disease, ruling out pulmonary or thoracic endometriosis may be necessary.[9]
Though fibrofolliculomas are unique to Birt–Hogg–Dubé, they may present with an ambiguous appearance and must be confirmed histologically. Other diseases can mimic the dermatologic manifestations of BHD, including tuberous sclerosis complex, Cowden syndrome, familial trichoepitheliomas, and multiple endocrine neoplasia type 1.[5] Tuberous sclerosis must be distinguished because both disorders can present with angiofibromas on the face, though they are more common in tuberous sclerosis.[6]
Management
The different manifestations of Birt–Hogg–Dubé syndrome are controlled in different ways. The fibrofolliculomas can be removed surgically, through curettage, shave excision, skin resurfacing, or laser ablation; however, this is not a permanent solution as the tumors often recur.[2][5] The renal and pulmonary symptoms are managed preventatively: CT scans, ultrasounds, or MRIs of the kidneys are recommended regularly, and family members are advised not to smoke.[2] MRIs are the preferred method for surveillance of the kidneys in people with BHD because they do not carry the same risk of radiation complications as CT scans and are more sensitive than ultrasounds.[6] Smokers with Birt–Hogg–Dubé have more severe pulmonary symptoms than non-smokers.[17][6] Though nephrectomy is sometimes indicated, kidney tumors in cases of Birt–Hogg–Dubé are often removed without taking the whole kidney, in a procedure called partial nephrectomy.[5] Knockout mouse studies have shown that administration of rapamycin may mitigate the effects of FLCN mutations on kidneys and improve renal cancer prognoses because of folliculin's interaction with the mTOR pathway.[6]
Epidemiology
The disorder has been reported in more than 100 families worldwide,[14] though some sources cite up to 400 families,[1] and it is inherited in an autosomal dominant pattern. It is considered to be under-diagnosed[3][6] because of the variability in its expression.[18] The pattern of mutations and spectrum of symptoms are heterogeneous between individuals.[14] Less severe skin phenotypes are seen in women and people of both sexes who have a late onset of skin symptoms.[5]
History
The syndrome was first well described in 1977,[19] by three Canadian physicians, Arthur R. Birt, Georgina R. Hogg and William J. Dubé. The earliest case of possible BHD in the medical literature was published by Burnier and Rejsek in 1927,[20] who described a case of perifollicular fibromas on a 56-year-old woman's face. Trichodiscomas were first described in 1974 by H.S. Zackheim and H. Pinkus, but were not associated with BHD until Birt, Hogg, and Dubé.[3] The first case of BHD with the systemic symptoms was described by Hornstein and Knickenberg and found in two siblings and their father, all of whom exhibited colon polyps and the characteristic fibrofolliculomas.[21] Though the siblings did not have renal or pulmonary symptoms, their father had cysts in his lungs and kidneys.[3] Hornstein-Knickenberg syndrome is a now-deprecated name for the inherited fibrofolliculomas inherent to Birt–Hogg–Dubé.[5]
Birt, Hogg, and Dubé examined a family with a hereditary thyroid cancer and discovered that many of the members had fibrofolliculomas, trichodiscomas, and acrochordons, which became defined as the classical symptoms of the eponymous disease. The first case of spontaneous pneumothorax associated with BHD was discovered in 1986;[3] the first case of renal cancer followed in 1993[6] and the presence of lung cysts in people with BHD was confirmed in 1999.[4] It was formerly thought that people with Birt–Hogg–Dubé syndrome were at higher risk for colorectal polyps and neoplasms, but this has been disproven.[2] The BHD Foundation supports research into the syndrome and holds regular symposia in BHD and related disorders for researchers, clinicians, and family members.[22][23][24]
Other animals
Genes related to FLCN and diseases similar to BHD have been found in dogs, fruit flies, rats, and mice. In German Shepherd dogs, missense mutations in the canine ortholog of FLCN cause a similar phenotype to human BHD—kidney cancers (in this case, mutifocal renal cystadenocarcinoma) and skin tumors (nodular dermatofibrosis). They had a similar pattern of tumorigenesis to human BHD in that the skin lesions were heterozygous for the FLCN mutation and the renal tumors were likely caused by loss of heterozygosity.[6] Female German shepherds with a FLCN mutation are also prone to uterine leiomyomas.[3]
A homolog of FLCN called DBHD has been discovered in the common fruit fly, Drosophila melanogaster.[25][3] Decrease expression of the DBHD results in loss of male germline stem cells (GSC), which suggest that DBHD is required for male GSC maintenance in the fly testis.[26] Further, DBHD regulates GSC maintenance downstream or in parallel of the JAK/STAT and Dpp signal-transduction pathways, which suggest that BHD regulates tumorigenesis by controlling stem cells in human {[27] Singh et al. 2006}
A line of rats with hereditary kidney cancer were developed by Japanese researchers. They have a mutation in the FLCN homolog that produces a truncated protein, though they do not develop the cutaneous or pulmonary symptoms seen in humans. Heterozygotes have renal abnormalities seen very early in life that develop into clear cell and hybrid tumors, significantly shortening the animals' lifespan; they also are prone to endometrial and salivary gland clear cell hyperplasia as well as rhabdomyolysis. Homozygotes do not survive to birth.[3] When a wild-type FLCN gene was added, the phenotype was rescued.[6]
Knockout mice have been created for a kidney-cancer causing mutation of BHD; heterozygotes develop kidney cysts and tumors that lead to renal failure within three weeks of birth. In these mice, the mTOR pathway was inappropriately activated, indicating that the mouse homolog of FLCN plays a regulatory role in this pathway. Rapamycin partially rescued the phenotype by regulating mTOR. Homozygotes die in utero.[3]
References
Citations
- ↑ 1.0 1.1 1.2 Genetics Home Reference.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 2.9 2.10 2.11 2.12 2.13 2.14 Andrews 2011.
- ↑ 3.0 3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 3.9 3.10 3.11 3.12 3.13 3.14 3.15 Reese et al. 2009.
- ↑ 4.0 4.1 4.2 4.3 4.4 4.5 Palmirotta et al. 2010.
- ↑ 5.0 5.1 5.2 5.3 5.4 5.5 5.6 5.7 5.8 5.9 5.10 5.11 5.12 5.13 5.14 5.15 5.16 5.17 5.18 5.19 5.20 5.21 5.22 5.23 5.24 Toro 2008.
- ↑ 6.0 6.1 6.2 6.3 6.4 6.5 6.6 6.7 6.8 6.9 6.10 6.11 6.12 6.13 6.14 6.15 6.16 6.17 Menko et al. 2009.
- ↑ 7.0 7.1 Chan-Smutko 2012, p. 345.
- ↑ 8.0 8.1 8.2 8.3 8.4 8.5 8.6 Coleman & Russo 2009, p. 482.
- ↑ 9.0 9.1 Furuya & Nakatani 2012.
- ↑ Grant, Babar & Griffin 2009, p. 442.
- ↑ Devine & Garcia 2012, p. 4.
- ↑ Coleman & Russo 2009, p. 481.
- ↑ Nickerson et al. 2002.
- ↑ 14.0 14.1 14.2 14.3 14.4 Toro et al. 2008.
- ↑ 15.0 15.1 Maher 2011.
- ↑ Sudarshan et al. 2013.
- ↑ Ayo et al. 2007.
- ↑ Verine et al. 2010.
- ↑ Birt, Hogg & Dubé 1977.
- ↑ Riegert-Johnson.
- ↑ Kniffin 2012.
- ↑ BHD Foundation.
- ↑ National Organization for Rare Disorders.
- ↑ Genetics Home Reference: Educational Resources.
- ↑ Liu et al. 2013.
- ↑ Singh SR, Zhen W, Zheng Z, Wang H, Oh SW, Liu W, Zbar B, Schmidt LS, Hou SX. The Drosophila homolog of the human tumor suppressor gene BHD interacts with the JAK-STAT and Dpp signaling pathways in regulating male germline stem cell maintenance. Oncogene. 2006 Sep 28;25(44):5933-41.
- ↑ Singh SR, Zhen W, Zheng Z, Wang H, Oh SW, Liu W, Zbar B, Schmidt LS, Hou SX. The Drosophila homolog of the human tumor suppressor gene BHD interacts with the JAK-STAT and Dpp signaling pathways in regulating male germline stem cell maintenance. Oncogene. 2006 Sep 28;25(44):5933-41
Bibliography
- Ayo, Dereje S.; Aughenbaugh, GL; Yi, ES; Hand, JL; Ryu, JH (2007), "Cystic Lung Disease in Birt-Hogg-Dubé Syndrome", CHEST Journal 132 (2): 679–84, doi:10.1378/chest.07-0042, PMID 17505035
- Birt, A. R.; Hogg, GR; Dubé, WJ (1977), "Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons", Archives of Dermatology 113 (12): 1674–7, doi:10.1001/archderm.113.12.1674, PMID 596896
- BHD Foundation, 2013, retrieved 17 July 2013
- Chan-Smutko, Gayun (2012), "Genetic Testing by Cancer Site", The Cancer Journal 18 (4): 343–9, doi:10.1097/PPO.0b013e31826246ac, PMID 22846736
- Coleman, Jonathan A; Russo, Paul (2009), "Hereditary and familial kidney cancer", Current Opinion in Urology 19 (5): 478–85, doi:10.1097/MOU.0b013e32832f0d40, PMID 19584731
- Devine, Megan Stuebner; Garcia, Christine Kim (2012), "Genetic Interstitial Lung Disease", Clinics in Chest Medicine 33 (1): 95–110, doi:10.1016/j.ccm.2011.11.001, PMC 3292740, PMID 22365249
- Furuya, M.; Nakatani, Y. (2012), "Birt-Hogg-Dube syndrome: Clinicopathological features of the lung", Journal of Clinical Pathology 66 (3): 178–86, doi:10.1136/jclinpath-2012-201200, PMC 3595143, PMID 23223565
- "Birt-Hogg-Dubé syndrome", Genetics Home Reference (US National Library of Medicine), January 2013, retrieved 13 July 2013
- "Birt-Hogg-Dubé syndrome: Educational resources", Genetics Home Reference (NIH), 22 July 2013, retrieved 25 July 2013
- Grant, L.A.; Babar, J.; Griffin, N. (2009), "Cysts, cavities, and honeycombing in multisystem disorders: Differential diagnosis and findings on thin-section CT", Clinical Radiology 64 (4): 439–48, doi:10.1016/j.crad.2008.09.015, PMID 19264190
- James, William D.; Berger, Timothy; Elston, Dirk (2011), Andrew's Diseases of the Skin: Clinical Dermatology (11th ed.), Elsevier Health Sciences, ISBN 978-1-4377-3619-9
- Kniffin, Cassandra L. (22 August 2012), "Birt-Hogg-Dube syndrome, BHD", Online Mendelian Inheritance in Man (Johns Hopkins University), retrieved 13 July 2013
- Liu, Wei; Chen, Zhi; Ma, Yansen; Wu, Xiaochun; Jin, Yaping; Hou, Steven (2013), White-Cooper, Helen, ed., "Genetic Characterization of the Drosophila Birt-Hogg-Dubé Syndrome Gene", PLoS ONE 8 (6): e65869, doi:10.1371/journal.pone.0065869, PMC 3684598, PMID 23799055
- Maher, Eamonn R. (2011), "Genetics of Familial Renal Cancers", Nephron Experimental Nephrology 118 (1): e21–6, doi:10.1159/000320892, PMID 21071978
- Menko, Fred H; Van Steensel, Maurice AM; Giraud, Sophie; Friis-Hansen, Lennart; Richard, Stéphane; Ungari, Silvana; Nordenskjöld, Magnus; Hansen, Thomas vO; Solly, John; Maher, Eamonn R; European Bhd, Consortium (2009), "Birt-Hogg-Dubé syndrome: Diagnosis and management", The Lancet Oncology 10 (12): 1199–206, doi:10.1016/S1470-2045(09)70188-3, PMID 19959076
- BHD Foundation, National Organization for Rare Disorders, 2013, retrieved 25 July 2013
- Nickerson, Michael L.; Warren, Michelle B.; Toro, Jorge R.; Matrosova, Vera; Glenn, Gladys; Turner, Maria L.; Duray, Paul; Merino, Maria; Choyke, Peter et al. (2002), "Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome", Cancer Cell 2 (2): 157–64, doi:10.1016/S1535-6108(02)00104-6, PMID 12204536
- Palmirotta, Raffaele; Savonarola, Annalisa; Ludovici, Giorgia; Donati, Pietro; Cavaliere, Francesco; De Marchis, Maria Laura; Ferroni, Patrizia; Guadagni, Fiorella (March 2010), "Association between Birt Hogg Dubé syndrome and cancer predisposition", Anticancer Res. 30 (3): 751–7, PMID 20392993
- Reese, Erin; Sluzevich, Jason; Kluijt, Irma; Teertstra, H. Jelle; De Jong, Daphne; Horenblas, Simon; Ryu, Jay (5 October 2009), "Birt-Hogg-Dubé Syndrome", in Riegert-Johnson, Douglas L; Boardman, Lisa A; Hefferon, Timothy; Roberts, Maegan, Cancer Syndromes, Bethesda, MD: National Center for Biotechnology Information, PMID 21249760
- Riegert-Johnson, DL, "Birt-Hogg-Dube", Familial Cancer Syndromes (NCBI), retrieved 21 July 2009
- Sudarshan, Sunil; Karam, Jose A.; Brugarolas, James; Thompson, R. Houston; Uzzo, Robert; Rini, Brian; Margulis, Vitaly; Patard, Jean-Jacques; Escudier, Bernard; Linehan, W. Marston (2013), "Metabolism of Kidney Cancer: From the Lab to Clinical Practice", European Urology 63 (2): 244–51, doi:10.1016/j.eururo.2012.09.054, PMC 3709870, PMID 23063455
- Toro, Jorge R. (September 9, 2008), "Birt-Hogg-Dubé Syndrome", in Pagon, Roberta A; Adam, Margaret P; Bird, Thomas D; Dolan, Cynthia R; Fong, Chin-To; Smith, Richard JH; Stephens, Karen, GeneReviews, University of Washington, PMID 20301695
- Toro, J R; Wei, M-H; Glenn, G M; Weinreich, M; Toure, O; Vocke, C; Turner, M; Choyke, P; Merino, M J et al. (2008), "BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: A new series of 50 families and a review of published reports", Journal of Medical Genetics 45 (6): 321–31, doi:10.1136/jmg.2007.054304, PMC 2564862, PMID 18234728
- Verine, Jérôme; Pluvinage, Amélie; Bousquet, Guilhem; Lehmann-Che, Jacqueline; De Bazelaire, Cédric; Soufir, Nadem; Mongiat-Artus, Pierre (2010), "Hereditary Renal Cancer Syndromes: An Update of a Systematic Review", European Urology 58 (5): 701–10, doi:10.1016/j.eururo.2010.08.031, PMID 20817385
External links
| Wikimedia Commons has media related to Birt–Hogg–Dubé syndrome. |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||