Biopython
|
| |
| Original author(s) | Chapman B, Chang J[1] |
|---|---|
| Initial release | 2000[1] |
| Stable release | 1.65 / 17 December 2014 |
| Development status | Active |
| Written in | Python and C |
| Platform | Cross-platform |
| Type | Bioinformatics |
| License | Biopython License |
| Website |
biopython |
The Biopython Project is an open-source collection of non-commercial Python tools for computational biology and bioinformatics, created by an international association of developers.[1][2][3] It contains classes to represent biological sequences and sequence annotations, and it is able to read and write to a variety of file formats. It also allows for a programmatic means of accessing online databases of biological information, such as those at NCBI. Separate modules extend Biopython's capabilities to sequence alignment, protein structure, population genetics, phylogenetics, sequence motifs, and machine learning. Biopython is one of a number of Bio* projects designed to reduce code duplication in computational biology.[4]
History
Biopython development began in 1999 and it was first released in July 2000.[5] It was developed during a similar time frame and with analogous goals to other projects that added bioinformatics capabilities to their respective programming languages, including BioPerl, BioRuby and BioJava. Early developers on the project included Jeff Chang, Andrew Dalke and Brad Chapman, though over 100 people have made contributions to date.[6] In 2007, a similar Python project, namely Py Cogent, was established.[7]
The initial scope of Biopython involved accessing, indexing and processing biological sequence files. While this is still a major focus, over the following years added modules have extended its functionality to cover additional areas of biology (see Key features and examples).
As of version 1.62, Biopython supports running on Python 3 as well as Python 2.[8]
Design
Wherever possible, Biopython follows the conventions used by the Python programming language to make it easier for users familiar with Python. For example, Seq and SeqRecord objects can be manipulated via slicing, in a manner similar to Python’s strings and lists. It is also designed to be functionally similar to other Bio* projects, such as BioPerl.[5]
Biopython is able to read and write most common file formats for each of its functional areas, and its license is permissive and compatible with most other software licenses, which allow Biopython to be used in a variety of software projects.[3]
Key features and examples
Sequences
A core concept in Biopython is the biological sequence, and this is represented by the Seq class.[9] A Biopython Seq object is similar to a Python string in many respects: it supports the Python slice notation, can be concatenated with other sequences and is immutable. In addition, it includes sequence-specific methods and specifies the particular biological alphabet used.
>>> # This script creates a DNA sequence and performs some typical manipulations >>> from Bio.Seq import Seq >>> from Bio.Alphabet import IUPAC >>> dna_sequence = Seq('AGGCTTCTCGTA', IUPAC.unambiguous_dna) >>> dna_sequence Seq('AGGCTTCTCGTA', IUPACUnambiguousDNA()) >>> dna_sequence[2:7] Seq('GCTTC', IUPACUnambiguousDNA()) >>> dna_sequence.reverse_complement() Seq('TACGAGAAGCCT', IUPACUnambiguousDNA()) >>> rna_sequence = dna_sequence.transcribe() >>> rna_sequence Seq('AGGCUUCUCGUA', IUPACUnambiguousRNA()) >>> rna_sequence.translate() Seq('RLLV', IUPACProtein())
Sequence annotation
The SeqRecord class describes sequences, along with information such as name, description and features in the form of SeqFeature objects. Each SeqFeature object specifies the type of the feature and its location. Feature types can be ‘gene’, ‘CDS’ (coding sequence), ‘repeat_region’, ‘mobile_element’ or others, and the position of features in the sequence can be exact or approximate.
>>> # This script loads an annotated sequence from file and views some of its contents. >>> from Bio import SeqIO >>> seq_record = SeqIO.read('pTC2.gb', 'genbank') >>> seq_record.name 'NC_019375' >>> seq_record.description 'Providencia stuartii plasmid pTC2, complete sequence.' >>> seq_record.features[14] SeqFeature(FeatureLocation(ExactPosition(4516), ExactPosition(5336), strand=1), type='mobile_element') >>> seq_record.seq Seq('GGATTGAATATAACCGACGTGACTGTTACATTTAGGTGGCTAAACCCGTCAAGC...GCC', IUPACAmbiguousDNA())
Input and output
Biopython can read and write to a number of common sequence formats, including FASTA, FASTQ, GenBank, Clustal, PHYLIP and NEXUS. When reading files, descriptive information in the file is used to populate the members of Biopython classes, such as SeqRecord. This allows records of one file format to be converted into others.
Very large sequence files can exceed a computer's memory resources, so Biopython provides various options for accessing records in large files. They can be loaded entirely into memory in Python data structures, such as lists or dictionaries, providing fast access at the cost of memory usage. Alternatively, the files can be read from disk as needed, with slower performance but lower memory requirements.
>>> # This script loads a file containing multiple sequences and saves each one in a different format. >>> from Bio import SeqIO >>> genomes = SeqIO.parse('salmonella.gb', 'genbank') >>> for genome in genomes: ... SeqIO.write(genome, genome.id + '.fasta', 'fasta')
Accessing online databases
Through the Bio.Entrez module, users of Biopython can download biological data from NCBI databases. Each of the functions provided by the Entrez search engine is available through functions in this module, including searching for and downloading records.
>>> # This script downloads genomes from the NCBI Nucleotide database and saves them in a FASTA file. >>> from Bio import Entrez >>> from Bio import SeqIO >>> output_file = open('all_records.fasta', "w") >>> Entrez.email = 'my_email@example.com' >>> records_to_download = ['FO834906.1', 'FO203501.1'] >>> for record_id in records_to_download: ... handle = Entrez.efetch(db='nucleotide', id=record_id, rettype='gb') ... seqRecord = SeqIO.read(handle, format='gb') ... handle.close() ... output_file.write(seqRecord.format('fasta'))
Phylogeny
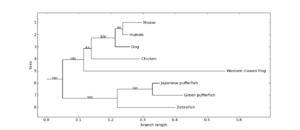
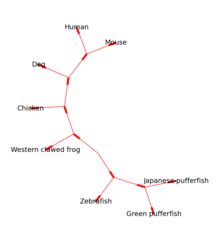
The Bio.Phylo module provides tools for working with and visualising phylogenetic trees. A variety of file formats are supported for reading and writing, including Newick, NEXUS and phyloXML. Common tree manipulations and traversals are supported via the Tree and Clade objects. Examples include converting and collating tree files, extracting subsets from a tree, changing a tree's root, and analysing branch features such as length or score.[11]
Rooted trees can be drawn in ASCII or using matplotlib (see Figure 1), and the Graphviz library can be used to create unrooted layouts (see Figure 2).
Genome diagrams
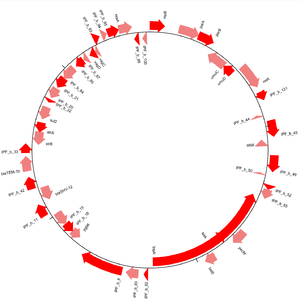
The GenomeDiagram module provides methods of visualising sequences within Biopython.[13] Sequences can be drawn in a linear or circular form (see Figure 3), and many output formats are supported, including PDF and PNG. Diagrams are created by making tracks and then adding sequence features to those tracks. By looping over a sequence's features and using their attributes to decide if and how they are added to the diagram's tracks, one can exercise much control over the appearance of the final diagram. Cross-links can be drawn between different tracks, allowing one to compare multiple sequences in a single diagram.
Macromolecular structure
The Bio.PDB module can load molecular structures from PDB and mmCIF files, and was added to Biopython in 2003.[14] The Structure object is central to this module, and it organises macromolecular structure in a hierarchical fashion: Structure objects contain Model objects which contain Chain objects which contain Residue objects which contain Atom objects. Disordered residues and atoms get their own classes, DisorderedResidue and DisorderedAtom, that describe their uncertain positions.
Using Bio.PDB, one can navigate through individual components of a macromolecular structure file, such as examining each atom in a protein. Common analyses can be carried out, such as measuring distances or angles, comparing residues and calculating residue depth.
Population genetics
The Bio.PopGen module adds support to Biopython for Genepop, a software package for statistical analysis of population genetics.[15] This allows for analyses of Hardy–Weinberg equilibrium, linkage disequilibrium and other features of a population's allele frequencies.
This module can also carry out population genetic simulations using coalescent theory with the fastsimcoal2 program.[16]
Wrappers for command line tools
Many of Biopython's modules contain command line wrappers for commonly used tools, allowing these tools to be used from within Biopython. These wrappers include BLAST, Clustal, PhyML, EMBOSS and SAMtools. Users can subclass a generic wrapper class to add support for any other command line tool.
See also
References
- ↑ 1.0 1.1 1.2 Chapman, Brad; Chang, Jeff (August 2000). "Biopython: Python tools for computational biology". ACM SIGBIO Newsletter 20 (2): 15–19. doi:10.1145/360262.360268.
- ↑ Cock, Peter JA; Antao, Tiago; Chang, Jeffery T; Chapman, Brad A; Cox, Cymon J; Dalke, Andrew; Friedberg, Iddo; Hamelryck, Thomas; Kauff, Frank; Wilczynski, Bartek; de Hoon, Michiel JL (20 March 2009). "Biopython: freely available Python tools for computational molecular biology and bioinformatics". Bioinformatics 25 (11): 1422–3. doi:10.1093/bioinformatics/btp163. PMC 2682512. PMID 19304878.
- ↑ 3.0 3.1 Refer to the Biopython website for other papers describing Biopython, and a list of over one hundred publications using/citing Biopython.
- ↑ Mangalam, Harry (September 2002). "The Bio* toolkits—a brief overview". Briefings in Bioinformatics 3 (3): 296–302. doi:10.1093/bib/3.3.296. PMID 12230038.
- ↑ 5.0 5.1 Chapman, Brad (11 March 2004), The Biopython Project: Philosophy, functionality and facts, retrieved 11 September 2014
- ↑ List of Biopython contributors, retrieved 11 September 2014
- ↑ Knight, R; Maxwell, P; Birmingham, A; Carnes, J; Caporaso, J. G.; Easton, B. C.; Eaton, M; Hamady, M; Lindsay, H; Liu, Z; Lozupone, C; McDonald, D; Robeson, M; Sammut, R; Smit, S; Wakefield, M. J.; Widmann, J; Wikman, S; Wilson, S; Ying, H; Huttley, G. A. (2007). "Py Cogent: A toolkit for making sense from sequence". Genome Biology 8 (8): R171. doi:10.1186/gb-2007-8-8-r171. PMC 2375001. PMID 17708774.
- ↑ Chapman, Brad, Biopython Installation, retrieved 11 September 2014
- ↑ Chang, Jeff; Chapman, Brad; Friedberg, Iddo; Hamelryck, Thomas; de Hoon, Michiel; Cock, Peter; Antao, Tiago; Talevich, Eric; Wilczynski, Bartek (29 May 2014), Biopython Tutorial and Cookbook, retrieved 28 August 2014
- ↑ Zmasek, Christian M; Zhang, Qing; Ye, Yuzhen; Godzik, Adam (24 October 2007). "Surprising complexity of the ancestral apoptosis network". Genome Biology 8 (10). doi:10.1186/gb-2007-8-10-r226. Retrieved 9 September 2014.
- ↑ Talevich, Eric; Invergo, Brandon M; Cock, Peter JA; Chapman, Brad A (21 August 2012). "Bio.Phylo: A unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython". BMC Bioinformatics 13 (209). doi:10.1186/1471-2105-13-209. Retrieved 9 September 2014.
- ↑ "Klebsiella pneumoniae strain KPS77 plasmid pKPS77, complete sequence". NCBI. Retrieved 10 September 2014.
- ↑ Pritchard, Leighton; White, Jennifer A; Birch, Paul RJ; Toth, Ian K (March 2006). "GenomeDiagram: a python package for the visualization of large-scale genomic data". Bioinformatics 22 (5): 616–617. doi:10.1093/bioinformatics/btk021.
- ↑ Hamelryck, Thomas; Manderick, Bernard (10 May 2003). "PDB file parser and structure class implemented in Python". Bioinformatics 19 (17). doi:10.1093/bioinformatics/btg299. Retrieved 10 September 2014.
- ↑ Rousset, François (January 2008). "GENEPOP'007: a complete re-implementation of the GENEPOP software for Windows and Linux". Molecular Ecology Resources 8. doi:10.1111/j.1471-8286.2007.01931.x. Retrieved 11 September 2014.
- ↑ Excoffier, Laurent; Foll, Matthieu (1 March 2011). "fastsimcoal: a continuous-time coalescent simulator of genomic diversity under arbitrarily complex evolutionary scenarios". Bioinformatics 27 (9). doi:10.1093/bioinformatics/btr124. Retrieved 11 September 2014.