BioJava
| Original author(s) | Andreas Prlić |
|---|---|
| Stable release | 3.0.5 |
| Preview release | Nightly builds |
| Development status | Active |
| Written in | Java |
| Platform | Cross-platform |
| Available in | Java |
| Type | Bioinformatics |
| License | Lesser GPL |
| Website |
biojava |
BioJava[1] is an open source project dedicated to providing Java tools for processing biological data.[2][3] BioJava is a set of library functions written in the Java programming language for manipulating sequences, protein structures, file parsers, CORBA interoperability, DAS, access to AceDB, dynamic programming, and simple statistical routines. BioJava supports a huge range of data, starting from DNA and protein sequences to the level of 3D protein structures. The BioJava libraries are useful for automating many daily and mundane bioinformatics tasks such as to parsing a PDB file, interacting with Jmol and many more.[4] This Application programming interface (API) provides various file parsers, data models and algorithms to facilitate working with the standard data formats and enables rapid application development and analysis. These libraries have also been used in many scientific studies. For example:
- MUSI: an integrated system for identifying multiple specificity from very large peptide or nucleic acid data sets.[5]
- JEnsembl: a version-aware Java API to Ensembl data systems.[6]
- Expression profiling of signature gene sets with trinucleotide threading[7]
- Resolving the structural features of genomic islands: a machine learning approach[8]
- Utility library for structural bioinformatics[9]
The BioJava project grew out of work by Thomas Down and Matthew Pocock to create an API to simplify development of Java based Bioinformatics tools. BioJava is an active open source project that has been developed over more than 12 years and by more than 60 developers. BioJava is one of a number of Bio* projects designed to reduce code duplication.[10] Examples of such projects that fall under Bio* apart from BioJava are BioPython,[11] BioPerl,[12] BioRuby,[13] EMBOSS[14] etc.
The latest version of BioJava (3.0.5) is a major update to the previous versions. The new version of BioJava contains several independent modules. The old project has been moved to a separate project called biojava-legacy project.
Features
BioJava provides software modules for many of the typical tasks of bioinformatics programming. These include:
- Accessing nucleotide and peptide sequence data from local and remote databases
- Transforming formats of database/ file records
- Protein structure parsing and manipulation
- Manipulating individual sequences
- Searching for similar sequences
- Creating and manipulating sequence alignments
History and Publications
In the year 2008, BioJava's first Application note was published.[2] It was migrated from its original CVS repository to Git hub on April 2013.[15]
In October 2012, the most recent paper on BioJava was published.[1] As of November 2012 Google Scholar counts more than 130 citations.
Modules
Over the last 2 years, large parts of the original code base have been rewritten. BioJava 3 is a clear departure from the version 1 series. It now consists of several independent modules built using an automation tool called Apache Maven.[16] These modules provide state-of-the-art tools for protein structure comparison, pairwise and multiple sequence alignments, working with DNA and protein sequences, analysis of amino acid properties, detection of protein modifications and prediction of disordered regions in proteins as well as parsers for common file formats using a biologically meaningful data model. The original code has been moved into a separate BioJava legacy project, which is still available for backwards compatibility.
The following sections will describe several of the new modules and highlight some of the new features that are included in the latest version of BioJava.
Core Module
This module provides Java classes to model amino acid or nucleotide sequences. The classes were designed keeping in mind that they should be familiar to the biologists i.e. the names should make sense to the biologists and at the same time also provide a concrete representation of the steps in going from a gene sequence to a protein sequence for the computer scientists and programmers.
A major change between the legacy BioJava project and BioJava3 lies in the way framework has been designed to take advantage of the recent innovations in Java. A sequence is defined as a generic interface allowing the rest of the modules to create any utility that operates on all sequences. Specific classes for common sequences such as DNA and proteins have been defined in order to improve usability for biologists. The translation engine really leverages this work by allowing conversions between DNA, RNA and amino acid sequences. This engine can handle details such as choosing the codon table, converting start codons to methionine, trimming stop codons, specifying the reading frame and handing ambiguous sequences.
Special attentions has been paid in designing the storage of sequences so as to minimize space requirements. Special design patterns such as the Proxy pattern allowed the developers to create the framework such that sequences can be stored in memory, fetched on demand from a web service such as UniProt or read from a FASTA file as needed. The latter two approaches save memory by not loading sequence data until it is referenced in the application. This concept can be extended to handle very large genomic datasets, such as NCBI GenBank or a proprietary database.
Protein structure modules
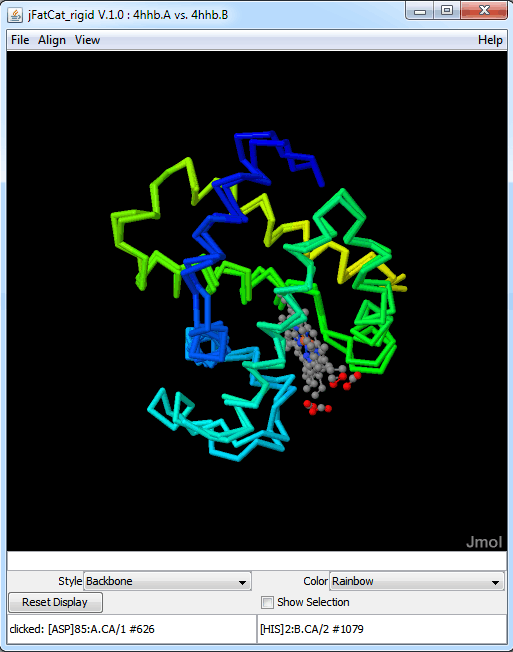
The protein structure modules provide tools for representing and manipulating 3D biomolecular structures. It particularly focuses on protein structure comparison.
The following algorithms have been implemented and included in BioJava.
- FATCAT algorithm for flexible and rigid body alignment.[17]
- The standard Combinatorial Extension (CE) algorithm.[18]
- A new version of CE that can detect circular permutations in proteins.[19]
These algorithms are used to provide the RCSB Protein Data Bank (PDB)[20] Protein Comparison Tool as well as systematic comparisons of all proteins in the PDB on a weekly basis.[21]
Parsers for PDB[22] and mmCIF[23] file formats allow the loading of structure data into a reusable data model. This feature is used by the SIFTS project to map between UniProt sequences and PDB structures.[24] Information from the RCSB PDB can be dynamically fetched without the need to manually download data. For visualization, an interface to the 3D viewer Jmol[4] http://www.jmol.org/ is provided. The team claims that work is underway to improve interaction with the RCSB PDB viewers.[25]
Below is an outline of the code to initialize a window that will display and compare two protein sequences. Please bear in mind that this is just an outline of the code. To make this work one will need to import the correct found in the "org.biojava.bio.structure" package and add also handle exceptions by using a try-catch block.
String name1 = "4hhb.A";
String name2 = "4hhb.B";
AtomCache cache = new AtomCache();
Structure structure1 = null;
Structure structure2 = null;
StructureAlignment algorithm =
StructureAlignmentFactory.getAlgorithm(FatCatRigid.algorithmName);
structure1 = cache.getStructure(name1);
structure2 = cache.getStructure(name2);
Atom[] ca1 = StructureTools.getAtomCAArray(structure1);
Atom[] ca2 = StructureTools.getAtomCAArray(structure2);
FatCatParameters params = new FatCatParameters();
AFPChain afpChain = algorithm.align(ca1,ca2,params);
afpChain.setName1(name1);
afpChain.setName2(name2);
StructureAlignmentDisplay.display(afpChain, ca1, ca2);
The code aligns the two protein sequences "4hhb.A" and "4hhb.B" based on the FATCAT rigid algorithm.[17]
Genome and Sequencing modules
This module is focused on the creation of gene sequence objects from the core module. This is realised by supporting the parsing of the following popular standard file formats generated by open source gene prediction applications:
- GTF files generated by GeneMark[26]
- GFF2 files generated by GeneID[27]
- GFF3 files generated by Glimmer[28]
Then the gene sequence objects are written out as a GFF3 format and is imported into GMOD.[29] These file formats are well defined but what gets written in the file is very flexible.
The following code example takes a 454scaffold file that was used by genemark to predict genes and returns a collection of ChromosomeSequences. Each chromosome sequence maps to a named entry in the fasta file and would contain N gene sequences. The gene sequences can be +/- strand with frame shifts and multiple transcriptions.
Passing the collection of ChromsomeSequences to GeneFeatureHelper.getProteinSequences would return all protein sequences. You can then write the protein sequences to a fasta file.
LinkedHashMap<String, ChromosomeSequence> chromosomeSequenceList = GeneFeatureHelper.loadFastaAddGeneFeaturesFromGeneMarkGTF(new File("454Scaffolds.fna"), new File("genemark_hmm.gtf")); LinkedHashMap<String, ProteinSequence> proteinSequenceList = GeneFeatureHelper.getProteinSequences(chromosomeSequenceList.values()); FastaWriterHelper.writeProteinSequence(new File("genemark_proteins.faa"), proteinSequenceList.values());
You can also output the gene sequence to a fasta file where the coding regions will be upper case and the non-coding regions will be lower case
LinkedHashMap<String, GeneSequence> geneSequenceHashMap = GeneFeatureHelper.getGeneSequences(chromosomeSequenceList.values()); Collection<GeneSequence> geneSequences = geneSequenceHashMap.values(); FastaWriterHelper.writeGeneSequence(new File("genemark_genes.fna"), geneSequences, true);
You can easily write out a gff3 view of a ChromosomeSequence with the following code.
FileOutputStream fo = new FileOutputStream("genemark.gff3"); GFF3Writer gff3Writer = new GFF3Writer(); gff3Writer.write(fo, chromosomeSequenceList); fo.close();
For providing input-output support for several common variants of the FASTQ file format from the next generation sequencers,[30] a separate sequencing module is provided. It is called the Sequence Module and is contained in the package org.biojava3.sequencing.io.fastq. For samples on how to use this module please go to this link.
Work is in progress towards providing a complete set of java classes to do conversions between different file formats where the list of supported gene prediction applications and genome browsers will get longer based on end user requests.
Alignment module
This module contains several classes and methods that allow users to perform pairwise and multiple sequence alignment.
Pairwise sequence alignment
For optimal global alignment, BioJava implements the Needleman-Wunsch[31] algorithm and for performing local alignments the Smith and Waterman's[32] algorithm has been implemented.
The outputs of both local and global alignments are available in standard formats.
An example on how to use the libraries is shown below.
protected void align(String uniProtID_1, String uniProtID_2, PairwiseSequenceAlignerType alignmentType) throws IOException, Exception { ProteinSequence proteinSeq1 = FastaReaderHelper.readFastaProteinSequence((new URL(String.format ("http://www.uniprot.org/uniprot/%s.fasta", uniProtID_1))).openStream()).get(uniProtID_1); ProteinSequence proteinSeq2 = FastaReaderHelper.readFastaProteinSequence((new URL(String.format ("http://www.uniprot.org/uniprot/%s.fasta", uniProtID_2))).openStream()).get(uniProtID_2); SequencePair<ProteinSequence, AminoAcidCompound> result = Alignments.getPairwiseAlignment(proteinSeq1, proteinSeq2, alignmentType, new SimpleGapPenalty(), new SimpleSubstitutionMatrix<AminoAcidCompound>()); System.out.println(result.toString()); }
An example call to the above function would look something like this:
For Global Alignment
align("Q21691", "Q21495", PairwiseSequenceAlignerType.GLOBAL);
For Local Alignment
align("Q21691", "Q21495", PairwiseSequenceAlignerType.LOCAL);
In addition to these two algorithms, there is an implementation of Guan–Uberbacher algorithm[33] which performs global sequence alignment very efficiently since it only uses linear memory.
For Multiple Sequence Alignment, any of the methods discussed above can be used to progressively perform a multiple sequence alignment.
ModFinder module

The ModFinder module provides new methods to identify and classify protein modifications in protein 3D structures. Over 400 different types of protein modifications such as phosphorylation, glycosylation, disulfide bonds metal chelation etc. were collected and curated based on annotations in PSI-MOD,[35] RESID[36] and RCSB PDB.[37] The module also provides an API for detecting protein modifications within protein structures.
Example: identify and print all preloaded modifications from a structure[38]
Set<ModifiedCompound> identifyAllModfications(Structure struc) { ProteinModificationIdentifier parser = new ProteinModificationIdentifier(); parser.identify(struc); Set<ModifiedCompound> mcs = parser.getIdentifiedModifiedCompound(); return mcs; }
Example: identify phosphorylation sites in a structure
List<ResidueNumber> identifyPhosphosites(Structure struc) { List<ResidueNumber> phosphosites = new ArrayList<ResidueNumber>(); ProteinModificationIdentifier parser = new ProteinModificationIdentifier(); parser.identify(struc, ProteinModificationRegistry.getByKeyword("phosphoprotein")); Set<ModifiedCompound> mcs = parser.getIdentifiedModifiedCompound(); for (ModifiedCompound mc : mcs) { Set<StructureGroup> groups = mc.getGroups(true); for (StructureGroup group : groups) { phosphosites.add(group.getPDBResidueNumber()); } } return phosphosites; }
Demo code to run the above methods
import org.biojava.bio.structure.ResidueNumber; import org.biojava.bio.structure.Structure; import org.biojava.bio.structure.io.PDBFileReader; import org.biojava3.protmod.structure.ProteinModificationIdentifier; public static void main(String[] args) { try { PDBFileReader reader = new PDBFileReader(); reader.setAutoFetch(true); // identify all modifications from PDB:1CAD and print them String pdbId = "1CAD"; Structure struc = reader.getStructureById(pdbId); Set<ModifiedCompound> mcs = identifyAllModfications(struc); for (ModifiedCompound mc : mcs) { System.out.println(mc.toString()); } // identify all phosphosites from PDB:3MVJ and print them pdbId = "3MVJ"; struc = reader.getStructureById(pdbId); List<ResidueNumber> psites = identifyPhosphosites(struc); for (ResidueNumber psite : psites) { System.out.println(psite.toString()); } } catch(Exception e) { e.printStackTrace(); } }
There are plans to include additional protein modifications by integrating other resources such as UniProt[39]
Amino acid properties module
This module attempts to provide accurate physio-chemical properties of proteins. The properties that can calculated using this module are as follows:
- Molecular mass
- Extinction coefficient
- Instability index
- Aliphatic index
- Grand average of hydropathy
- Isoelectric point
- Amino acid composition
The precise molecular weights for common isotopically labelled amino acids are included in this module. There also exists flexibility to define new amino acid molecules with their molecular weights using simple XML configuration files. This can be useful where the precise mass is of high importance such as mass spectrometry experiments.
Protein disorder module
The goal of this module is to provide users ways to find disorders in protein molecules. BioJava includes a Java implementation of the RONN predictor. The latest version of BioJava(3.0.5) makes use of Java's support for multithreading to improve performance by up to 3.2 times,[40] on a modern quad-core machine, as compared to the legacy C implementation.
There are two ways to use this module:
- Using library function calls
- Using command line
Making library function calls
The following examples show how to use the module and make function calls to get information about protein disorders. The first two examples make library function calls to calculate the probability of disorder for every residue in the sequence provided.
The third and fourth examples demonstrates how easily one can get the disordered regions of the protein.
Example 1: Calculate the probability of disorder for every residue in the sequence
FastaSequence fsequence = new FastaSequence("name", "LLRGRHLMNGTMIMRPWNFLNDHHFPKFFPHLIEQQAIWLADWWRKKHC" + "RPLPTRAPTMDQWDHFALIQKHWTANLWFLTFPFNDKWGWIWFLKDWTPGSADQAQRACTWFFCHGHDTN"); float[] rawProbabilityScores = Jronn.getDisorderScores(fsequence);
Example 2: Calculate the probability of disorder for every residue in the sequence for all proteins from the FASTA input file
final List<FastaSequence> sequences = SequenceUtil.readFasta(new FileInputStream("src/test/resources/fasta.in")); Map<FastaSequence, float[]> rawProbabilityScores = Jronn.getDisorderScores(sequences);
Example 3: Get the disordered regions of the protein for a single protein sequence
FastaSequence fsequence = new FastaSequence("Prot1", "LLRGRHLMNGTMIMRPWNFLNDHHFPKFFPHLIEQQAIWLADWWRKKHC" + "RPLPTRAPTMDQWDHFALIQKHWTANLWFLTFPFNDKWGWIWFLKDWTPGSADQAQRACTWFFCHGHDTN" + "CQIIFEGRNAPERADPMWTGGLNKHIIARGHFFQSNKFHFLERKFCEMAEIERPNFTCRTLDCQKFPWDDP"); Range[] ranges = Jronn.getDisorder(fsequence);
Example 4: Calculate the disordered regions for the proteins from FASTA file
final List<FastaSequence> sequences = SequenceUtil.readFasta(new FileInputStream("src/test/resources/fasta.in")); Map<FastaSequence, Range[]> ranges = Jronn.getDisorder(sequences);
Using command line
BioJava module biojava3-protein-disorder can be compiled into a single executable JAR file and run using the following command.[41]
java -jar <jar_file_name>
Options supported by the command line executable
JRONN version 3.1b usage 1 August 2011: java -jar JRONN_JAR_NAME -i=inputfile <OPTIONS> Where -i=input file Input file can contain one or more FASTA formatted sequences. All OPTIONS are optional OPTION DETAILED DESCRIPTION: -o full path to the output file, if not specified standard out is used -d the value of disorder, defaults to 0.5 -f output format, V for vertical, where the letters of the sequence and corresponding disorder values are output in two column layout. H for horizontal, where the disorder values are provided under the letters of the sequence. Letters and values separated by tabulation in this case. Defaults to V. -s the file name to write execution statistics to. -n the number of threads to use. Defaults to the number of cores available on the computer. n=1 mean sequential processing. Valid values are 1 < n < (2 x num_of_cores) Default value will give the best performance.
Examples
Predict disorder values for sequences from input file /home/input.fasta output the results to the standard out. Use default disorder value and utilise all CPUs available on the computer.
java -jar JRONN.JAR -i=/home/input.fasta
Predict disorder values for sequences from input file /home/input.fasta output the results in horizontal layout to the /home/jronn.out, collect execution statistics to /home/jronn.stat.txt file and limit the number of threads to two.
java -jar JRONN.JAR -i=/home/input.fasta -o=/home/jronn.out -d=0.6 -n=2 -f=H
The arguments can be provided in any order.
Web service access module
As per the current trends in bioinformatics, web based tools are gaining popularity. The web service module allows bioinformatics services to be accessed using REST protocols. Currently, two services are implemented: NCBI Blast through the Blast URLAPI (previously known as QBlast) and the HMMER web service.[42]
Comparisons with other alternatives
The need for customized software in the field of bioinformatics has been addressed by several groups and individuals. Similar to BioJava, open source projects such as BioPerl, BioPython and BioRuby all provide tool-kits with multiple functionality that make it easier to create customized pipelines or analysis.
As the names suggest, the projects mentioned above use different programming languages. All of these APIs offer similar tools so on what criteria should one base their choice? For programmers who are experienced in only one of these languages, the choice is straightforward. However, for a well-rounded bioinformaticist who knows all of these languages and wants to choose the best language for a job the choice can be made based on the following guidelines given by a software review[10] done on the Bio* tool-kits.
In general, for small programs (<500 lines) that will be used only by a small group or individual, it is hard to beat Perl and BioPerl. These constraints probably cover the needs of 90 per cent of personal bioinformatics programming requirements.
For beginners, and for writing larger programs in the Bio domain, especially those to be shared and supported by others, Python’s clarity and brevity make it very attractive.
For those who might be leaning towards a career in bioinformatics and who want to learn only one language, Java has the widest general programming support, very good support in the Bio domain with BioJava, and is now the de facto language of business (the new COBOL, for better or worse).
Apart from these Bio* projects there is another project called STRAP which uses Java and aims for similar goals. The STRAP-toolbox, similar to BioJava is also a Java-toolkit for the design of Bioinformatics programs and scripts. The similarities and differences between BioJava and STRAP are as follows:
Similarities
- Both provide comprehensive collections of methods for protein sequences.
- Both are used by Java programmers for coding Bioinformatics algorithms.
- Both separate implementations and definitions by using java interfaces.
- Both are open source projects.
- Both can read and write many sequence file formats.
Differences
- BioJava is applicable to nucleotide and peptide sequences and can be applied for entire genomes. STRAP cannot cope with single sequences as long as an entire chromosome. Instead STRAP manipulates peptide sequences and 3D- structures of the size of single proteins. Nevertheless, it can hold a high number of sequences and structures in memory. STRAP is designed for protein sequences but can read coding nucleotide files, which are then translated to peptide sequences.
- STRAP is very fast since the graphical user interface must be highly responsive. BioJava is used where speed is less critical.
- BioJava is well designed in terms of type safety, ontology and object design. BioJava uses objects for sequences, annotations and sequence positions. Even single amino acids or nucleotides are object references. To enhance speed, STRAP avoids frequent object instantiations and invocation of non-final object-methods.
- In BioJava peptide sequences and nucleotide sequences are lists of symbols. The symbols can be retrieved one after the other with an iterator or sub-sequences can be obtained. The advantages are that the entire sequence does not necessarily reside in memory and that programs are less susceptible to programming errors. Symbol objects are immutable elements of an alphabet. In STRAP however simple byte arrays are used for sequences and float arrays for coordinates. Besides speed the low memory consumption is an important advantage of basic data types. Classes in Strap expose internal data. Therefore programmers might commit programming errors like manipulating byte arrays directly instead of using the setter methods. Another disadvantage is that no checks are performed in STRAP whether the characters in sequences are valid with respect to an underlying alphabet.
- In BioJava sequence positions are realized by the class Location. Discontiguous Location objects are composed of several contiguous RangeLocation objects or PointLocation objects. For the class StrapProtein however, single residue positions are indicated by integer numbers between 0 and countResidues()-1. Multiple positions are given by boolean arrays. True at a given index means selected whereas false means not selected.
- BioJava throws exceptions when methods are invoked with invalid parameters. STRAP avoids the time consuming creation of Throwable objects. Instead, errors in methods are indicated by the return values NaN, -1 or null. From the point of program design however Throwable objects are nicer.
- In BioJava a Sequence object is either a peptide sequence or a nucleotide sequence. A StrapProtein can hold both at the same time if a coding nucleotide sequence was read and translated into protein. Both, the nucleotide sequence and the peptide sequence are contained in the same StrapProtein object. The coding or non-coding regions can be changed and the peptide sequence alters accordingly.
Projects using BioJava
The following projects make use of BioJava.
- Metabolic Pathway Builder: Software suite dedicated to the exploration of connections among genes, proteins, reactions and metabolic pathways
- DengueInfo: a Dengue genome information portal that uses BioJava in the middleware and talks to a biosql database.
- Dazzle: A BioJava based DAS server.
- BioSense: A plugin for the InforSense Suite, an analytics software platform by IDBS that unitizes BioJava.
- Bioclipse: A free, open source, workbench for chemo- and bioinformatics with powerful editing and visualization capabilities for molecules, sequences, proteins, spectra etc.
- PROMPT: A free, open source framework and application for the comparison and mapping of protein sets. Uses BioJava for handling most input data formats.
- Cytoscape: An open source bioinformatics software platform for visualizing molecular interaction networks.
- BioWeka: An open source biological data mining application.
- Geneious: A molecular biology toolkit.
- MassSieve: An open source application to analyze mass spec proteomics data.
- Strap: A tool for multiple sequence alignment and sequence based structure alignment.
- Jstacs: A Java framework for statistical analysis and classification of biological sequences
- jLSTM "Long Short-Term Memory" for protein classification
- LaJolla Structural alignment of RNA and proteins using an index structure for fast alignment of thousands of structures. Including an easy to use command line interface. Open source at Sourceforge.
- GenBeans: A rich client platform for bioinformatics primarily focused on molecular biology and sequence analysis.
See also
External links
References
- ↑ 1.0 1.1 Prlić A, Yates A, Bliven SE et al. (October 2012). "BioJava: an open-source framework for bioinformatics in 2012". Bioinformatics 28 (20): 2693–5. doi:10.1093/bioinformatics/bts494. PMC 3467744. PMID 22877863.
- ↑ 2.0 2.1 Holland RC, Down TA, Pocock M, Prlić A, Huen D, James K et al. (2008). "BioJava: an open-source framework for bioinformatics.". Bioinformatics 24 (18): 2096–7. doi:10.1093/bioinformatics/btn397. PMC 2530884. PMID 18689808.
- ↑ VS Matha and P Kangueane, 2009, Bioinformatics: a concept-based introduction, 2009. p26
- ↑ 4.0 4.1 4.2 Hanson,R.M. (2010) Jmol a paradigm shift in crystallographic visualization.
- ↑ Kim T, Tyndel MS, Huang H et al. (March 2012). "MUSI: an integrated system for identifying multiple specificity from very large peptide or nucleic acid data sets". Nucleic Acids Res. 40 (6): e47. doi:10.1093/nar/gkr1294. PMC 3315295. PMID 22210894.
- ↑ Paterson T, Law A (November 2012). "JEnsembl: a version-aware Java API to Ensembl data systems". Bioinformatics 28 (21): 2724–31. doi:10.1093/bioinformatics/bts525. PMC 3476335. PMID 22945789.
- ↑ Zajac P, Pettersson E, Gry M, Lundeberg J, Ahmadian A (February 2008). "Expression profiling of signature gene sets with trinucleotide threading". Genomics 91 (2): 209–17. doi:10.1016/j.ygeno.2007.10.012. PMID 18061398.
- ↑ Vernikos GS, Parkhill J (February 2008). "Resolving the structural features of genomic islands: a machine learning approach". Genome Res. 18 (2): 331–42. doi:10.1101/gr.7004508. PMC 2203631. PMID 18071028.
- ↑ Gront D, Kolinski A (February 2008). "Utility library for structural bioinformatics". Bioinformatics 24 (4): 584–5. doi:10.1093/bioinformatics/btm627. PMID 18227118.
- ↑ 10.0 10.1 Mangalam H (2002). "The Bio* toolkits--a brief overview.". BRIEFINGS IN BIOINFORMATICS 3 (3): 296–302. doi:10.1093/bib/3.3.296. PMID 12230038.
- ↑ Cock PJ, Antao T, Chang JT et al. (June 2009). "Biopython: freely available Python tools for computational molecular biology and bioinformatics". Bioinformatics 25 (11): 1422–3. doi:10.1093/bioinformatics/btp163. PMC 2682512. PMID 19304878.
- ↑ Stajich JE, Block D, Boulez K et al. (October 2002). "The Bioperl toolkit: Perl modules for the life sciences". Genome Res. 12 (10): 1611–8. doi:10.1101/gr.361602. PMC 187536. PMID 12368254.
- ↑ Goto N, Prins P, Nakao M, Bonnal R, Aerts J, Katayama T (October 2010). "BioRuby: bioinformatics software for the Ruby programming language". Bioinformatics 26 (20): 2617–9. doi:10.1093/bioinformatics/btq475. PMC 2951089. PMID 20739307.
- ↑ Rice P, Longden I, Bleasby A (June 2000). "EMBOSS: the European Molecular Biology Open Software Suite". Trends Genet. 16 (6): 276–7. doi:10.1016/S0168-9525(00)02024-2. PMID 10827456.
- ↑ "History". Retrieved 30 Jan 2015.
- ↑ Maven, Apache. "Maven". Apache.
- ↑ 17.0 17.1 17.2 Ye Y, Godzik A (October 2003). "Flexible structure alignment by chaining aligned fragment pairs allowing twists". Bioinformatics 19 (Suppl 2): ii246–55. doi:10.1093/bioinformatics/btg1086. PMID 14534198.
- ↑ Shindyalov IN, Bourne PE (September 1998). "Protein structure alignment by incremental combinatorial extension (CE) of the optimal path". Protein Eng. 11 (9): 739–47. doi:10.1093/protein/11.9.739. PMID 9796821.
- ↑ Bliven S, Prlić A (2012). "Circular permutation in proteins". PLoS Comput. Biol. 8 (3): e1002445. doi:10.1371/journal.pcbi.1002445. PMC 3320104. PMID 22496628.
- ↑ Rose PW, Beran B, Bi C et al. (January 2011). "The RCSB Protein Data Bank: redesigned web site and web services". Nucleic Acids Res. 39 (Database issue): D392–401. doi:10.1093/nar/gkq1021. PMC 3013649. PMID 21036868.
- ↑ Prlic A, Bliven S, Rose PW et al. (December 2010). "Pre-calculated protein structure alignments at the RCSB PDB website". Bioinformatics 26 (23): 2983–5. doi:10.1093/bioinformatics/btq572. PMC 3003546. PMID 20937596.
- ↑ Bernstein FC, Koetzle TF, Williams GJ et al. (May 1977). "The Protein Data Bank: a computer-based archival file for macromolecular structures". J. Mol. Biol. 112 (3): 535–42. doi:10.1016/s0022-2836(77)80200-3. PMID 875032.
- ↑ Fitzgerald,P.M.D. et al. (2006) Macromolecular dictionary (mmCIF). In Hall,S.R.
- ↑ Velankar S, McNeil P, Mittard-Runte V et al. (January 2005). "E-MSD: an integrated data resource for bioinformatics". Nucleic Acids Res. 33 (Database issue): D262–5. doi:10.1093/nar/gki058. PMC 540012. PMID 15608192.
- ↑ Moreland JL, Gramada A, Buzko OV, Zhang Q, Bourne PE (2005). "The Molecular Biology Toolkit (MBT): a modular platform for developing molecular visualization applications". BMC Bioinformatics 6: 21. doi:10.1186/1471-2105-6-21. PMC 548701. PMID 15694009.
- ↑ Besemer J, Borodovsky M (July 2005). "GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses". Nucleic Acids Res. 33 (Web Server issue): W451–4. doi:10.1093/nar/gki487. PMC 1160247. PMID 15980510.
- ↑ Blanco E, Abril JF (2009). "Computational gene annotation in new genome assemblies using GeneID". Methods Mol. Biol. 537: 243–61. doi:10.1007/978-1-59745-251-9_12. PMID 19378148.
- ↑ Kelley DR, Liu B, Delcher AL, Pop M, Salzberg SL (January 2012). "Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering". Nucleic Acids Res. 40 (1): e9. doi:10.1093/nar/gkr1067. PMC 3245904. PMID 22102569.
- ↑ Stein LD, Mungall C, Shu S et al. (October 2002). "The generic genome browser: a building block for a model organism system database". Genome Res. 12 (10): 1599–610. doi:10.1101/gr.403602. PMC 187535. PMID 12368253.
- ↑ Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM (April 2010). "The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants". Nucleic Acids Res. 38 (6): 1767–71. doi:10.1093/nar/gkp1137. PMC 2847217. PMID 20015970.
- ↑ Needleman SB, Wunsch CD (March 1970). "A general method applicable to the search for similarities in the amino acid sequence of two proteins". J. Mol. Biol. 48 (3): 443–53. doi:10.1016/0022-2836(70)90057-4. PMID 5420325.
- ↑ Smith TF, Waterman MS (March 1981). "Identification of common molecular subsequences". J. Mol. Biol. 147 (1): 195–7. doi:10.1016/0022-2836(81)90087-5. PMID 7265238.
- ↑ Guan X, Uberbacher EC (February 1996). "Alignments of DNA and protein sequences containing frameshift errors". Comput. Appl. Biosci. 12 (1): 31–40. doi:10.1093/bioinformatics/12.1.31. PMID 8670617.
- ↑ Chen K, Jung YS, Bonagura CA et al. (February 2002). "Azotobacter vinelandii ferredoxin I: a sequence and structure comparison approach to alteration of [4Fe-4S]2+/+ reduction potential". J. Biol. Chem. 277 (7): 5603–10. doi:10.1074/jbc.M108916200. PMID 11704670.
- ↑ Montecchi-Palazzi L, Beavis R, Binz PA et al. (August 2008). "The PSI-MOD community standard for representation of protein modification data". Nat. Biotechnol. 26 (8): 864–6. doi:10.1038/nbt0808-864. PMID 18688235.
- ↑ Garavelli JS (June 2004). "The RESID Database of Protein Modifications as a resource and annotation tool". Proteomics 4 (6): 1527–33. doi:10.1002/pmic.200300777. PMID 15174122.
- ↑ Berman HM, Westbrook J, Feng Z et al. (January 2000). "The Protein Data Bank". Nucleic Acids Res. 28 (1): 235–42. doi:10.1093/nar/28.1.235. PMC 102472. PMID 10592235.
- ↑ CookBook3, BioJava. "ModFinder".
- ↑ Farriol-Mathis N, Garavelli JS, Boeckmann B et al. (June 2004). "Annotation of post-translational modifications in the Swiss-Prot knowledge base". Proteomics 4 (6): 1537–50. doi:10.1002/pmic.200300764. PMID 15174124.
- ↑ Yang ZR, Thomson R, McNeil P, Esnouf RM (August 2005). "RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins". Bioinformatics 21 (16): 3369–76. doi:10.1093/bioinformatics/bti534. PMID 15947016.
- ↑ CookBook3, BioJava. "Protein Disorder CLI".
- ↑ Finn RD, Clements J, Eddy SR (July 2011). "HMMER web server: interactive sequence similarity searching". Nucleic Acids Res. 39 (Web Server issue): W29–37. doi:10.1093/nar/gkr367. PMC 3125773. PMID 21593126.