Azomethine ylide

Azomethine ylides are nitrogen-based 1,3-dipoles, consisting of an iminium ion next to a carbanion. They are used in 1,3-dipolar cycloaddition reactions to form 5-membered heterocycles, including pyrrolidines and pyrrolines.[1][2][3] These reactions are highly stereo- and regioselective, and have the potential to form four new contiguous stereocenters. Azomethine ylides thus have high utility in total synthesis, and formation of chiral ligands and pharmaceuticals. Azomethine ylides can be generated from many sources, including aziridines, imines, and iminiums. They are often generated in situ, and immediately reacted with dipolarophiles.
Structure
The resonance structures below show the 1,3-dipole contribution, in which the two carbon atoms adjacent to the nitrogen have a negative or positive charge.[1] The most common representation of azomethine ylides is that in which the nitrogen is positively charged, and the negative charge is shared between the two carbons atoms. The relative contributions of the different resonance structures depend on the substituents on each atom. The carbon containing electron-withdrawing substituents will have a more partial negative charge, due to the ability of the nearby electron-withdrawing group to stabilize the negative charge.

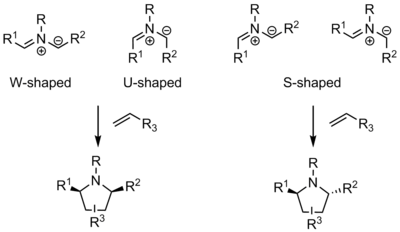
Three different ylide shapes are possible, each leading to different stereochemistry in the products of 1,3-dipolar cycloaddition reactions. W-shaped, U-shaped, and S-shaped ylides are possible.[1] The W- and U-shaped ylides, in which the R substituents are on the same side, result in syn cycloaddition products, whereas S-shaped ylides result in anti products. In the examples below, where the R3 substituent ends up in the product depends on the substituent's steric and electronic nature (see regioselectivity of 1,3 dipolar cycloadditions). The stereochemistry of R1 and R2 in the cycloaddition product is derived from the dipole. The stereochemistry of R3 is derived from the dipolarophile—-if the dipolarophile is more than mono-substituted (and prochiral), up to four new stereocenters can result in the product.
Generation
From aziridines
Azomethine ylides can be generated from ring opening of aziridines.[4][5] In accordance with the Woodward–Hoffmann rules, the thermal 4-electron ring opening proceeds via a conrotatory process, whereas the photochemical reaction is disrotatory.

In this ring opening reaction, there is an issue of torquoselectivity. Electronegative substituents prefer to rotate outwards, to the same side as the R substituent on the nitrogen, whereas electropositive substituents prefer to rotate inwards.[6]
Note that with aziridines, ring opening can result in a different 1,3-dipole, in which a C-N bond (rather than the C-C bond) breaks.[7]
By condensation of aldehyde with amine
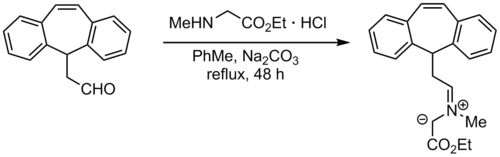
One of the easiest methods of forming azomethine ylides is by condensation of an aldehyde with an amine. If the amine contains an electron-withdrawing group on the alpha carbon, such as an ester, the deprotonation occurs readily. A possible disadvantage of using this method is that the ester ends up in the cycloaddition product. An alternative is to use a carboxylic acid, which can easily be removed during the cycloaddition process by decarboxylation.[8]
From imines and iminiums

Azomethine ylides can also be formed directly by deprotonation of iminiums.
By N-Metallation
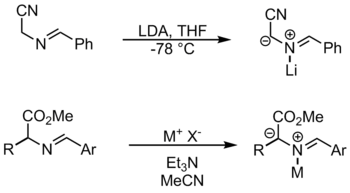
The metals used in this reaction include LiBr and AgOAc.[1] In this method, the metal coordinates to the N in order to activate the substrate for deprotonation. Another way to form azomethine ylides from imines is by prototropy and by alkylation.
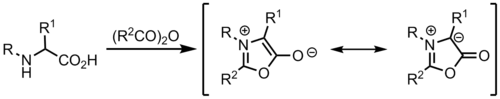
From münchnones
Ylides can be formed from münchnones, which are mesoionic heterocycles, and act as cyclic azomethine ylides.[9]
1,3-dipolar cycloaddition reactions

As with other cycloaddition reactions of a 1,3-dipole with a π-system, 1,3-dipolar cycloaddition using an azomethine ylide is a 6-electron process. According to the Woodward-Hoffman rules, this addition is suprafacial with respect to both the dipole and dipolarophile. The reaction is generally viewed as concerted, in which the two carbon-carbon bonds are being formed at the same time, but asynchronously. However, depending on the nature of the dipole and dipolarophile, diradical or zwitterionic intermediates are possible.[10] The endo product is generally favored, as in the isoelectronic Diels–Alder reaction. In these reactions, the azomethine ylide is typically the HOMO, and the electron-deficient dipolarophile the LUMO, although cycloaddition reactions with unactivated π-systems are known to occur, especially when the cyclization is intramolecular.[11] For a discussion of frontier molecular orbital theory of 1,3-dipolar cycloadditions, see 1,3-dipolar cycloaddition#Frontier molecular orbital theory.
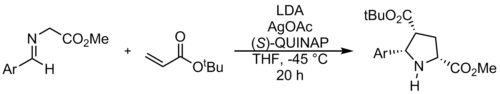
1,3-dipolar cycloaddition reactions of azomethine ylides commonly use alkenes or alkynes as dipolarophiles, to form pyrrolidines or pyrrolines, respectively. A reaction of an azomethine ylide with an alkene is shown above, and results in a pyrrolidine.[12] While dipolarophiles are typically α,β-unsaturated carbonyl compounds, there have been many recent advances in developing new types of dipolarophiles.[13]
When the dipole and dipolarophile are part of the same molecule, an intramolecular cyclization reaction can lead to a polycyclic product of considerable complexity.[1] If the dipolarophile is tethered to a carbon of the dipole, a fused bicycle is formed. If it is tethered to the nitrogen, a bridged structure results. The intramolecular nature of the reaction can also be useful in that regioselectivity is often constrained. Another advantage to intramolecular reactions is that the dipolarophile need not be electron-deficient—-many examples of cyclization reactions with electron-rich, alkyl-substituted dipolarophiles have been reported, including the synthesis of martinellic acid shown below.
Stereoselectivity of cycloadditions
Unlike most 1,3-dipolar cycloaddition reactions, in which the stereochemistry of the dipole is lost or non-existent, azomethine ylides are able to retain their stereochemistry. This is generally done by ring opening of an aziridine, and subsequent trapping by a dipolarophile before the stereochemistry can scramble.
Like other 1,3-dipolar cycloaddition reactions, azomethine ylide cycloadditions can form endo or exo products. This selectivity can be tuned using metal catalysis.[14][15]
Enantioselective synthesis
Enantioselective cycloaddition of azomethine ylides using chiral catalysts was first described in a seminal work by Allway and Grigg in 1991.[16] This powerful method was further developed by Jørgensen and Zhang. These reactions generally use Zn, Ag, Cu, Ni, and Ca complexes. Using chiral phosphine catalysts, enantiomerically pure spiroindolinones can be synthesized. The method described by Gong, et al. leads to an unexpected regiochemical outcome that does not follow electronic effects. This is attributed to favorable pi stacking with the catalyst.[17]
Other reactions
Electrocyclizations
Conjugated azomethine ylides are capable of [1,5]- and [1,7]-electrocyclizations.[18] An example of a [1,7]-electrocyclization of a diphenylethenyl-substituted azomethine ylide is shown below. This conrotatory ring-closing is followed by a suprafacial [1,5]-hydride shift, which affords the rearomatized product. The sterics and geometry of the reacting phenyl ring play a major role in the success of the reaction.[19]

The compounds resulting from this type of electrocyclization have been used as dienes in Diels–Alder reactions to attach compounds to fullerenes.[20]
Use in synthesis
Total synthesis of martinellic acid
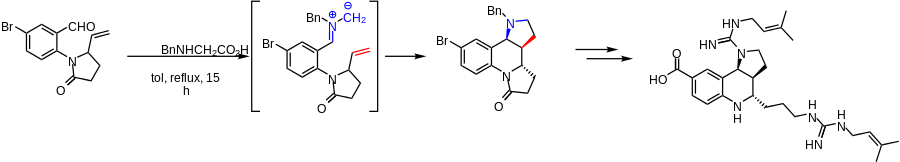
A cycloaddition of an azomethine ylide with an unactivated alkene was used in total synthesis of martinellic acid. The cycloaddition step formed two rings, including a pyrrolidine, and two stereocenters.[21]
Total synthesis of spirotryprostatin A

In the synthesis of spirotryprostatin A, an azomethine ylide is formed from condensation of an amine with an aldehyde. The ylide then reacts with an electron-deficient alkene on an indolinone, resulting in formation of a spirocyclic pyrrolidine and four contiguous stereocenters.[22]
Synthesis of benzodiazepinones
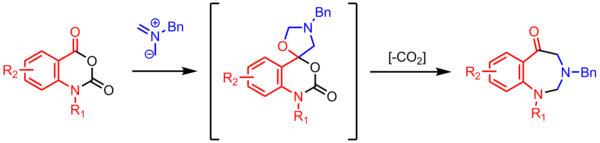
Cyclization of an azomethine ylide with a carbonyl affords a spirocyclic oxazolidine, which loses CO2 to form a 7-membered ring. These high-utility decarboxylative multi-step reactions are common in azomethine ylide chemistry.[23]
References
- ↑ 1.0 1.1 1.2 1.3 1.4 Coldham, Iain; Hufton, Richard (2005). "Intramolecular Dipolar Cycloaddition Reactions of Azomethine Ylides". Chemical Reviews 105 (7): 2765–2809. doi:10.1021/cr040004c.
- ↑ Padwa, Albert; Pearson, William H.; Harwood, L. M.; Vickers, R. J. (2003). "Chapter 3. Azomethine Ylides". Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products 59. doi:10.1002/0471221902.ch3.
- ↑ Adrio, Javier; Carretero, Juan C. (2011). "Novel dipolarophiles and dipoles in the metal-catalyzed enantioselective 1,3-dipolar cycloaddition of azomethine ylides". Chemical Communications 47 (24): 6784–6794. doi:10.1039/c1cc10779h.
- ↑ Dauban, Philippe; Malik Guillaume (2009). "A Masked 1,3-Dipole Revealed from Aziridines". Angewandte Chemie International Edition 48 (48): 9026–9029. doi:10.1002/anie.200904941.
- ↑ Huber, Helmut (1967). "Stereospecific Conversion of cis-trans Isomeric Aziridines to Open-Chain Azomethine Ylides". Journal of the American Chemical Society 89: 1753–1755. doi:10.1021/ja00983a052.
- ↑ Banks, Harold D. (2010). "Torquoselectivity Studies in the Generation of Azomethine Ylides from Substituted Aziridines". Journal of Organic Chemistry 75 (8): 2510–2517. doi:10.1021/jo902600y.
- ↑ Cardoso, Ana, L.; Pinho e Melo, Teresa M. V. D. (2012). "Aziridines in Formal [3+2] Cycloadditions: Synthesis of Five-Membered Heterocycles". European Journal of Organic Chemistry (33): 6479–6501. doi:10.1002/ejoc.201200406.
- ↑ Huie, Edward (1983). "Intramolecular [3 + 2] cycloaddition routes to carbon-bridged dibenzocycloheptanes and dibenzazepines". Journal of Organic Chemistry 48 (18): 2994–2997. doi:10.1021/jo00166a011.
- ↑ Padwa, Albert; Gingrich, Henry L.; Lim, Richard (1982). "Regiochemistry of intramolecular munchnone cycloadditions: preparative and mechanistic implications". Journal of Organic Chemistry 47 (12): 2447–56. doi:10.1021/jo00133a041.
- ↑ Li, Yi; Houk, Kendall N.; Gonzalez, Javier (1995). "Pericyclic Reaction Transition States". Acc. Chem. Res. 20 (2): 81–90. doi:10.1021/ar00050a004.
- ↑ Heathcock, Clayton H.; Henke, Brad R.; Kouklis, Andrew J. (1992). "Intramolecular 1,3-Dipolar Cycloaddition of Stabilized Azomethine Ylides to Unactivated Dipolarophiles". Journal of Organic Chemistry 57 (56): 7056–66. doi:10.1021/jo00052a015.
- ↑ Streiber, SL (2003). "Catalytic asymmetric [3+2] cycloaddition of azomethine ylides. Development of a versatile stepwise, three-component reaction for diversity-oriented synthesis.". J Am Chem Soc. 125 (34): 10174–5. doi:10.1021/ja036558z.
- ↑ Adrio, Javier; Carreter, Juan C. (2011). "Novel dipolarophiles and dipoles in the metal-catalyzed enantioselective 1,3-dipolar cycloaddition of azomethine ylides". Chem. Commun. 47: 6784–6794. doi:10.1039/c1cc10779h.
- ↑ Zhang, Xumu; Malati Raghunath; Wenzhong Gao (2005). "Cu(I)-Catalyzed Highly Exo-selective and Enantioselective [3 + 2] Cycloaddition of Azomethine Ylides with Acrylates". Organic Letters 7 (19): 4241–4244. doi:10.1021/ol0516925.
- ↑ Fukuzawa, Shin-ichi; Ichiro Oura; Kenta Shimizu; Kenichi Ogata (2010). "Highly Endo-Selective and Enantioselective 1,3-Dipolar Cycloaddition of Azomethine Ylide with α-Enones Catalyzed by a Silver(I)/ThioClickFerrophos Complex". Organic Letters 12 (8): 1752–1755. doi:10.1021/ol100336q.
- ↑ Allway, Philip; Grigg, Ronald (1991). "Chiral cobalt(II) and manganese(II) catalysts for the 1,3-dipolar cycloaddition reactions of azomethine ylides derived from arylidene imines of glycine". Tetrahedron Letters 32 (41): 5817–20. doi:10.1016/S0040-4039(00)93563-9.
- ↑ Gong, Liu-Zhu; Chen, Xiao-Hua; Wei, Qiang; Luo, Shi-Wei; Xiao, Han (2009). "Organocatalytic Synthesis of Spiro[pyrrolidin-3,3′-oxindoles] with High Enantiopurity and Structural Diversity". J. Am. Chem. Soc. 131 (38): 13819–13825. doi:10.1021/ja905302f.
- ↑ Nedolya, N. A.; Trofimov, B. A. (2013). "[1,7]-Electrocyclization reactions in the synthesis of azepine derivatives". Chemistry of Heterocyclic Compounds 49 (1): 152–176. doi:10.1007/s10593-013-1236-y.
- ↑ Nyerges, Miklós (2006). "1,7-Electrocyclization reactions of stabilized α,β:γ,δ-unsaturated azomethine ylides". Tetrahedron 16 (24): 5725–5735. doi:10.1016/j.tet.2006.03.088.
- ↑ Nierengarten, Jean-François (2002). "An unexpected Diels–Alder reaction on the fullerene core rather than an expected 1,3-dipolar cycloaddition". Chem. Commun. (7): 712–713. doi:10.1039/B201122K.
- ↑ Snider, BB; Ahn Y; O'Hare SM (2001). "Total synthesis of (+/-)-martinellic acid". Organic Letters 3 (26): 4217–20. doi:10.1021/ol016884o.
- ↑ Williams, Robert (2003). "Concise, Asymmetric Total Synthesis of Spirotryprostatin A". Organic Letters 5 (17): 3135–37. doi:10.1021/ol0351910.
- ↑ Ryan, John H (2011). "1,3-Dipolar cycloaddition-decarboxylation reactions of an azomethine ylide with isatoic anhydrides: formation of novel benzodiazepinones". Organic Letters 13 (3): 486–489. doi:10.1021/ol102824k.