Alignment-free sequence analysis
The emergence and need for the analysis of different types of data generated through biological research has given rise to the field of bioinformatics.[1] Molecular sequence and structure data of DNA, RNA and proteins, gene expression profiles or micro array data, metabolic pathway data are some of the major types of data being analysed in bioinformatics. Among them sequence data is increasing at the exponential rate due to advent of next-generation sequencing technologies. Since the origin of bioinformatics, sequence analysis has remained the major area of research with wide range of applications in database searching, genome annotation, comparative genomics, molecular phylogeny and gene prediction. The pioneering approaches for sequence analysis were based on sequence alignment either global or local, pairwise or multiple sequence alignment.[2][3] Alignment-based approaches generally give excellent results when the sequences under study are closely related and can be reliably aligned, but when the sequences are divergent, a reliable alignment cannot be obtained and hence the applications of sequence alignment are limited. Another limitation of alignment-based approaches is their computational complexity and are time-consuming and thus, are limited when dealing with large-scale sequence data.[4] The advent of next generation sequencing technologies has resulted in generation of voluminous sequencing data. The size of this sequence data poses challenges on alignment-based algorithms in their assembly, annotation and comparative studies. Thus, alignment-free sequence analysis approaches provide attractive alternatives over alignment-based approaches.[5]
Alignment-free methods
Alignment-free methods can broadly be classified into four categories: a) methods based on k-mer/word frequency, b) methods based on substrings, c) methods based on information theory and d) methods based on graphical representation. Alignment-free approaches have been used in sequence similarity searches,[6] clustering and classification of sequences,[7] and more recently in phylogenetics[8][9] (Figure 1).
Such molecular phylogeny analyses employing alignment-free approaches are said to be part of next-generation phylogenomics.[9] A number of review articles provide in-depth review of alignment-free methods in sequence analysis.[10][11][12][13]
Methods based on k-mer/word frequency
The popular methods based on k-mer/word frequencies include feature frequency profile (FFP),[14][15] Composition vector (CV),[16][17] Return time distribution (RTD),[18][19] frequency chaos game representation (FCGR).[20] and Spaced Words [21]
Feature frequency profile (FFP)
The methodology involved in FFP based method starts by calculating the count of each possible k-mer (possible number of k-mers for nucleotide sequence: 4k, while that for protein sequence: 20k) in sequences. Each k-mer count in each sequence is then normalized by dividing it by total of all k-mers' count in that sequence. This leads to conversion of each sequence into its feature frequency profile. The pair wise distance between two sequences is then calculated Jensen-Shannon (JS) divergence between their respective FFPs. The distance matrix thus obtained can be used to construct phylogenetic tree using clustering algorithms like Neighbor-joining, UPGMA etc.
Composition vector (CV)
In this method frequency of appearance of each possible k-mer in a given sequence is calculated. The next characteristic step of this method is the subtraction of random background of these frequencies using Markov model to reduce the influence of random neutral mutations to highlight the role of selective evolution. The normalized frequencies are put a fixed order to form the composition vector (CV) of a given sequence. Cosine distance function is then used to compute pairwise distance between CVs of sequences. The distance matrix thus obtained can be used to construct phylogenetic tree using clustering algorithms like Neighbor-joining, UPGMA etc. This method can be extended through resort to efficient pattern matching algorithms to include in the computation of the composition vectors: (i) all k-mers for any value of k, (ii) all substrings of any length up to an arbitrarily set maximum k value, (iii) all maximal substrings, where a substring is maximal if extending it by any character would cause a decrease in its occurrence count .[22][23]
Return time distribution (RTD)
The RTD based method does not calculate the count of k-mers in sequences, instead it computes the time required for the reappearance of k-mers. The time refers to the number of residues in successive appearance of particular k-mer. Thus the occurrence of each k-mer in a sequence is calculated in the form of RTD, which is then summarised using two statistical parameters mean (μ) and standard deviation (σ). Thus each sequence is represented in the form of numeric vector of size 2*4k containing μ and σ of 4k RTDs. The pair wise distance between sequences is calculated using Euclidean distance measure. The distance matrix thus obtained can be used to construct phylogenetic tree using clustering algorithms like Neighbor-joining, UPGMA etc.
Frequency chaos game representation (FCGR)
The FCGR methods have evolved from Chaos game representation (CGR) technique, which provides scale independent representation for genomic sequences.[24] The CGRs can be divided by grid lines where each grid square denotes the occurrence of oligonucleotides of a specific length in the sequence. Such representation of CGRs is termed as Frequency Chaos Game Representation (FCGR). This leads to representation of each sequence into FCGR. The pair wise distance between FCGRs of sequences can be calculated using either the Pearson distance or the Euclidean distance.[25]
Spaced-word frequencies
While most alignment-free algorithms compare the word-composition of sequences, Spaced Words uses a pattern of care and don't care positions. The occurrence of a spaced word in a sequence is then defined by the characters at the match positions only, while the characters at the don't care positions are ignored. Instead of comparing the frequencies of contiguous words in the input sequences, this approach compares the frequencies of the spaced words according to the pre-defined pattern.[21]
Methods based on substrings
The methods in this category employ the similarity and differences of substrings in a pair of sequences. These algorithms were mostly used for string processing in computer science.[26]
Average common substring (ACS)
In this approach, for a chosen pair of sequences (A and B of lengths l and m respectively), longest substring starting at some position is identified in one sequence (A) which exactly matches in the other sequence (B) at any position. In this way, lengths of longest substrings starting at different positions in sequence A and having exact matches at some positions in sequence B are calculated. All these lengths are averaged to derive a measure 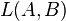 . Intuitively, larger the , the more similar the two sequences are. To account for the differences in the length of sequences, is normalized [i.e.
. Intuitively, larger the , the more similar the two sequences are. To account for the differences in the length of sequences, is normalized [i.e. 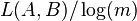 ]. This gives the similarity measure between the sequences.
]. This gives the similarity measure between the sequences.
In order to derive a distance measure, the inverse of similarity measure is taken and a correction term is subtracted from it to assure that 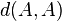 will be zero.
will be zero.
Thus, ![d(A, B) = [\log(m)/L(A, B)] - [\log(n)/L(A, A]](../I/m/0af713a6209fd56869d8cb175410c61a.png) .
.
This measure 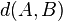 is not symmetric, so one has to compute
is not symmetric, so one has to compute 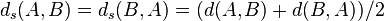 , which gives final ACS measure between the two strings (A and B).[27] The subsequence/substring search can be efficiently performed by
using suffix trees.[28][29][30]
, which gives final ACS measure between the two strings (A and B).[27] The subsequence/substring search can be efficiently performed by
using suffix trees.[28][29][30]
k-mismatch average common substring approach (kmacs)
This approach is a generalization of the ACS approach. To define the distance between two DNA or protein sequences, kmacs estimates for each position i of the first sequence the longest substring starting at i and matching a substring of the second sequence with up to k mismatches. It defines the average of these values as a measure of similarity between the sequences and turns this into a symmetric distance measure. Kmacs does not compute exact k-mismatch substrings, since this would be computational too costly, but approximates such substrings.[31]
Mutation distances (Kr)
This approach is closely related to the ACS, which calculates the number of substitutions per site between two DNA sequences using the shortest absent substring (termed as shustring).[32]
Methods based on Information theory
Information Theory has provided successful methods for alignment-free sequence analysis and comparison. The existing applications of information theory include global and local characterization of DNA, RNA and proteins, estimating genome entropy to motif and region classification. It also holds promise in gene mapping, next-generation sequencing analysis and metagenomics.[33]
Base base correlation (BBC)
Base base correlation (BBC)converts the genome sequence into a unique 16-dimensional numeric vector using the following equation,
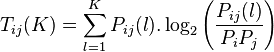
The 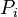 and
and 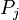 denotes the probabilities of bases i and j in the genome. The
denotes the probabilities of bases i and j in the genome. The 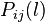 indicates the probability of bases i and j at distance l in the genome. The parameter K indicates the maximum distance between the bases i and j. The variation in the values of 16 parameters reflect variation in the genome content and length.[34][35][36]
indicates the probability of bases i and j at distance l in the genome. The parameter K indicates the maximum distance between the bases i and j. The variation in the values of 16 parameters reflect variation in the genome content and length.[34][35][36]
Information correlation and partial information correlation (IC-PIC)
IC-PIC (information correlation and partial information correlation) based method employs the base correlation property of DNA sequence. IC and PIC were calculated using following formulas,
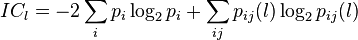
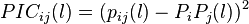
The final vector is obtained as following,
 here
here 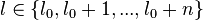 which defines the range of distance between bases.[37]
which defines the range of distance between bases.[37]
The pair wise distance between sequences is calculated using Euclidean distance measure. The distance matrix thus obtained can be used to construct phylogenetic tree using clustering algorithms like Neighbor-joining, UPGMA etc.
Lempel-Ziv compress
Lempel-Ziv complexity uses the relative information between the sequences. This complexity is measured by the number of steps required to generate a string given the prior knowledge of another string and a self-delimiting production process. This measure has a relation to measuring k-words in a sequence, as they can be easily used to generate the sequence. It is computational intensive method. Otu and Sayood (2003) used this method to construct five different distance measures for phylogenetic tree construction.[38]
Methods based on graphical representation
Iterated Maps
The use of iterated maps for sequence analysis was first introduced by HJ Jefferey in 1990[24] when he proposed to apply the Chaos Game to map genomic sequences into a unit square. That report coined the procedure as Chaos Game Representation (CGR). However, only 3 years later this approach was first dismissed as a projection of a Markov transition table by N Goldman.[39] This objection was overruled by the end of that decade when the opposite was found to be the case - that CGR bijectively maps Markov transition is into a fractal, order-free (degree-free) representation.[40] The realization that iterated maps provide a bijective map between the symbolic space and numeric space led to the identification of a variety of alignment-free approaches to sequence comparison and characterization. These developments were reviewed in late 2013 by JS Almeida in.[41] A number of web apps such as http://usm.github.com are available to demonstrate how to encode and compare arbitrary symbolic sequences.
Comparison of alignment based and alignment-free methods [9]
| Alignment-based methods | Alignment-free methods |
|---|---|
| These methods assume that homologous regions are contiguous (with gaps) | Does not assume such contiguity of homologous regions |
| Computes all possible pairwise comparisons of sequences; hence computationally expensive | Based on occurrences of sub-sequences; composition; computationally inexpensive, can be memory-intensive |
| Well-established approach in phylogenomics | Relatively recent and application in phylogenomics is limited; needs further testing for robustness and scalability |
| Requires substitution/evolutionary models | Less dependent on substitution/evolutionary models |
| Sensitive to stochastic sequence variation, recombination, horizontal (or lateral) genetic transfer, rate heterogeneity and sequences of varied lengths, especially when similarity lies in the “twilight zone” | Less sensitive to stochastic sequence variation, recombination, horizontal (or lateral) genetic transfer, rate heterogeneity and sequences of varied lengths |
| Best practice uses inference algorithms with complexity at least O(n2); less time-efficient | Inference algorithms typically O(n2) or less; more time-efficient |
| Heuristic in nature; statistical significance of how alignment scores relate to homology is difficult to assess | Exact solutions; statistical significance of the sequence distances (and degree of similarity) can be readily assessed |
Applications of alignment-free methods
- Molecular phylogenetics[9]
- Metagenomics[42]
- Next generation sequence data analysis[42]
- Epigenomics[43]
- Barcoding of species[44]
- Population genetics[45]
- Horizontal gene transfer[46]
- Sero/genotyping of viruses[18][19][47]
- Allergenicity prediction[48]
- SNP discovery[49]
- Recombination detection[50]
List of web servers/software for alignment-free methods
| Name | Description | Availability | Reference |
|---|---|---|---|
| kmacs | k-mismatch average common substring approach | kmacs | [31] |
| Spaced words | Spaced-word frequencies | spaced-words | [21] |
| FFP | Feature frequency profile based phylogeny | FFP | [14] |
| CVTree | Composition vector based server for phylogeny | CVTree | [51] |
| RTD Phylogeny | Return time distribution based server for phylogeny | RTD Phylogeny | [18] |
| AGP | A multimethods web server for alignment-free genome phylogeny | AGP | [52] |
| Alfy | Alignment-free detection of local similarity among viral and bacterial genomes | Alfy | [46] |
| decaf+py | DistancE Calculation using Alignment-Free methods in PYthon | decaf+py | [53] |
| MuV genotyping server | Genotyping of Mumps viruses based on RTD | MuV Genotyping server | [19] |
| Dengue Subtyper | Genotyping of Dengue viruses based on RTD | Dengue Subtyper | [18] |
| WNV Typer | Genotyping of West nile viruses based on RTD | WNV Typer | [47] |
| AllergenFP | Allergenicity prediction by descriptor fingerprints | AllergenFP | [48] |
| kSNP v2 | Alignment-Free SNP Discovery | kSNP v2 | [49] |
| d2Tools | Comparison of Metatranscriptomic Samples Based on k-Tuple Frequencies | d2Tools | [54] |
| rush | Recombination detection Using SHustrings | rush | [50] |
See also
- Sequence analysis
- Multiple sequence alignment
- Phylogenomics
- Bioinformatics
- Metagenomics
- Next-generation sequencing
- Population genetics
- SNPs
- Recombination detection program
References
- ↑ Rothberg, J; Merriman, B; Higgs, G (September 2012). "Bioinformatics. Introduction". The Yale journal of biology and medicine 85 (3): 305–8. PMC 3447194. PMID 23189382.
- ↑ Batzoglou, S (March 2005). "The many faces of sequence alignment". Briefings in bioinformatics 6 (1): 6–22. doi:10.1093/bib/6.1.6. PMID 15826353.
- ↑ Mullan, L (March 2006). "Pairwise sequence alignment--it's all about us!". Briefings in bioinformatics 7 (1): 113–5. doi:10.1093/bib/bbk008. PMID 16761368.
- ↑ Kemena, C; Notredame, C (Oct 1, 2009). "Upcoming challenges for multiple sequence alignment methods in the high-throughput era". Bioinformatics (Oxford, England) 25 (19): 2455–65. doi:10.1093/bioinformatics/btp452. PMC 2752613. PMID 19648142.
- ↑ Vinga, S; Almeida, J (Mar 1, 2003). "Alignment-free sequence comparison-a review.". Bioinformatics (Oxford, England) 19 (4): 513–23. doi:10.1093/bioinformatics/btg005. PMID 12611807.
- ↑ Hide, W; Burke, J; Davison, DB (1994). "Biological evaluation of d2, an algorithm for high-performance sequence comparison.". Journal of Computational Biology 1 (3): 199–215. doi:10.1089/cmb.1994.1.199. PMID 8790465.
- ↑ Miller, RT; Christoffels, AG; Gopalakrishnan, C; Burke, J; Ptitsyn, AA; Broveak, TR; Hide, WA (1999). "A comprehensive approach to clustering of expressed human gene sequence: the sequence tag alignment and consensus knowledge base.". Genome Research 9 (11): 1143–55. doi:10.1101/gr.9.11.1143. PMID 10568754.
- ↑ Domazet-Lošo, M; Haubold, B (2011). "Alignment-free detection of local similarity among viral and bacterial genomes.". Bioinformatics 27 (11): 1466–72. doi:10.1093/bioinformatics/btr176. PMID 21471011.
- ↑ 9.0 9.1 9.2 9.3 Chan, CX; Ragan, MA (Jan 22, 2013). "Next-generation phylogenomics.". Biology direct 8: 3. doi:10.1186/1745-6150-8-3. PMID 23339707.
- ↑ Vinga, S; Almeida, J (2003). "Alignment-free sequence comparison-a review.". Bioinformatics 19 (4): 513–23. doi:10.1093/bioinformatics/btg005. PMID 12611807.
- ↑ Song, K; Ren, J; Reinert, G; Deng, M; Waterman, MS; Sun, F (2014). "New developments of alignment-free sequence comparison: measures, statistics and next-generation sequencing.". Briefings in Bioinformatics 15 (3): 343–53. doi:10.1093/bib/bbt067. PMID 24064230.
- ↑ Haubold, B (2014). "Alignment-free phylogenetics and population genetics.". Briefings in Bioinformatics 15 (3): 407–18. doi:10.1093/bib/bbt083. PMID 24291823.
- ↑ Bonham-Carter, O; Steele, J; Bastola, D (2013). "Alignment-free genetic sequence comparisons: a review of recent approaches by word analysis.". Briefings in Bioinformatics 15: Epub ahead of print. doi:10.1093/bib/bbt052. PMID 23904502.
- ↑ 14.0 14.1 Sims, GE; Jun, SR; Wu, GA; Kim, SH (Oct 6, 2009). "Whole-genome phylogeny of mammals: evolutionary information in genic and nongenic regions.". Proceedings of the National Academy of Sciences of the United States of America 106 (40): 17077–82. doi:10.1073/pnas.0909377106. PMID 19805074.
- ↑ Sims, GE; Kim, SH (May 17, 2011). "Whole-genome phylogeny of Escherichia coli/Shigella group by feature frequency profiles (FFPs).". Proceedings of the National Academy of Sciences of the United States of America 108 (20): 8329–34. doi:10.1073/pnas.1105168108. PMC 3100984. PMID 21536867.
- ↑ Gao, L; Qi, J (Mar 15, 2007). "Whole genome molecular phylogeny of large dsDNA viruses using composition vector method.". BMC Evolutionary Biology 7: 41. doi:10.1186/1471-2148-7-41. PMID 17359548.
- ↑ Wang, H; Xu, Z; Gao, L; Hao, B (Aug 10, 2009). "A fungal phylogeny based on 82 complete genomes using the composition vector method.". BMC Evolutionary Biology 9: 195. doi:10.1186/1471-2148-9-195. PMC 3087519. PMID 19664262.
- ↑ 18.0 18.1 18.2 18.3 Kolekar, P; Kale, M; Kulkarni-Kale, U (November 2012). "Alignment-free distance measure based on return time distribution for sequence analysis: applications to clustering, molecular phylogeny and subtyping.". Molecular Phylogenetics & Evolution 65 (2): 510–22. doi:10.1016/j.ympev.2012.07.003. PMID 22820020.
- ↑ 19.0 19.1 19.2 Kolekar, PS; Kale, M; Kulkarni-Kale, U (Nov 30, 2011). "Genotyping of Mumps viruses based on SH gene: Development of a server using alignment-free and alignment-based methods.". Immunome research 7 (3): 1–7. PMID 22126822.
- ↑ Hatje, K; Kollmar, M (2012). "A phylogenetic analysis of the brassicales clade based on an alignment-free sequence comparison method.". Frontiers in plant science 3: 192. doi:10.3389/fpls.2012.00192. PMID 22952468.
- ↑ 21.0 21.1 21.2 Leimeister, CA; Boden, M; Horwege, S; Lindner, S (2014). "Fast alignment-free sequence comparison using spaced-word frequencies". Bioinformatics 30 (14): 1991–1999. doi:10.1093/bioinformatics/btu177.
- ↑ Apostolico, A; Denas, O (March 2008). "Fast algorithms for computing sequence distances by exhaustive substring composition.". Algorithms for Molecular Biology 3.
- ↑ Apostolico, A; Denas, O; Dress, A (September 2010). "Efficient tools for comparative substring analysis.". Journal of Biotechnology 149 (3): 120–126. doi:10.1016/j.jbiotec.2010.05.006.
- ↑ 24.0 24.1 Jeffrey, HJ (Apr 25, 1990). "Chaos game representation of gene structure.". Nucleic Acids Research 18 (8): 2163–70. doi:10.1093/nar/18.8.2163. PMID 2336393.
- ↑ Wang, Y; Hill, K; Singh, S; Kari, L (Feb 14, 2005). "The spectrum of genomic signatures: from dinucleotides to chaos game representation.". Gene 346: 173–85. doi:10.1016/j.gene.2004.10.021. PMID 15716010.
- ↑ Gusfield, Dan (1997). Algorithms on strings, trees, and sequences : computer science and computational biology (Reprinted (with corr.) ed.). Cambridge [u.a.]: Cambridge Univ. Press. ISBN 9780521585194.
- ↑ Ulitsky, I; Burstein, D; Tuller, T; Chor, B (March 2006). "The average common substring approach to phylogenomic reconstruction.". Journal of computational biology : a journal of computational molecular cell biology 13 (2): 336–50. doi:10.1089/cmb.2006.13.336. PMID 16597244.
- ↑ Weiner, P. "Linear pattern matching algorithms". IEEE.
- ↑ He, D (2006). "Using suffix tree to discover complex repetitive patterns in DNA sequences.". Conference proceedings : ... Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Society. Conference 1: 3474–7. doi:10.1109/IEMBS.2006.260445. PMID 17945779.
- ↑ Välimäki, N; Gerlach, W; Dixit, K; Mäkinen, V (Mar 1, 2007). "Compressed suffix tree--a basis for genome-scale sequence analysis.". Bioinformatics (Oxford, England) 23 (5): 629–30. doi:10.1093/bioinformatics/btl681. PMID 17237063.
- ↑ 31.0 31.1 Leimeister, CA; Morgenstern, B (2014). "kmacs: the k-Mismatch Average Common Substring Approach to alignment-free sequence comparison". Bioinformatics 30 (14): 2000–2008. doi:10.1093/bioinformatics/btu331.
- ↑ Haubold, B; Pfaffelhuber, P; Domazet-Loso, M; Wiehe, T (October 2009). "Estimating mutation distances from unaligned genomes.". Journal of computational biology : a journal of computational molecular cell biology 16 (10): 1487–500. doi:10.1089/cmb.2009.0106. PMID 19803738.
- ↑ Vinga, S (Sep 20, 2013). "Information theory applications for biological sequence analysis.". Briefings in bioinformatics 15: 376–389. doi:10.1093/bib/bbt068. PMID 24058049.
- ↑ Liu, Z; Meng, J; Sun, X (Apr 4, 2008). "A novel feature-based method for whole genome phylogenetic analysis without alignment: application to HEV genotyping and subtyping.". Biochemical and Biophysical Research Communications 368 (2): 223–30. doi:10.1016/j.bbrc.2008.01.070. PMID 18230342.
- ↑ Liu, ZH; Sun, X (2008). "Coronavirus phylogeny based on base-base correlation.". International journal of bioinformatics research and applications 4 (2): 211–20. doi:10.1504/ijbra.2008.018347. PMID 18490264.
- ↑ Cheng, J; Zeng, X; Ren, G; Liu, Z (Mar 14, 2013). "CGAP: a new comprehensive platform for the comparative analysis of chloroplast genomes.". BMC Bioinformatics 14: 95. doi:10.1186/1471-2105-14-95. PMID 23496817.
- ↑ Gao, Y; Luo, L (Jan 15, 2012). "Genome-based phylogeny of dsDNA viruses by a novel alignment-free method.". Gene 492 (1): 309–14. doi:10.1016/j.gene.2011.11.004. PMID 22100880.
- ↑ Otu, HH; Sayood, K (Nov 1, 2003). "A new sequence distance measure for phylogenetic tree construction.". Bioinformatics (Oxford, England) 19 (16): 2122–30. doi:10.1093/bioinformatics/btg295. PMID 14594718.
- ↑ Goldman, N (May 25, 1993). "Nucleotide, dinucleotide and trinucleotide frequencies explain patterns observed in chaos game representations of DNA sequences.". Nucleic Acids Research 21 (10): 2487–91. doi:10.1093/nar/21.10.2487. PMID 8506142.
- ↑ Almeida, JS; Carriço, JA; Maretzek, A; Noble, PA; Fletcher, M (May 2001). "Analysis of genomic sequences by Chaos Game Representation.". Bioinformatics (Oxford, England) 17 (5): 429–37. doi:10.1093/bioinformatics/17.5.429. PMID 11331237.
- ↑ Almeida, JS (Oct 25, 2013). "Sequence analysis by iterated maps, a review.". Briefings in bioinformatics 15: 369–375. doi:10.1093/bib/bbt072. PMID 24162172.
- ↑ 42.0 42.1 Song, K; Ren, J; Reinert, G; Deng, M; Waterman, MS; Sun, F (Nov 26, 2013). "New developments of alignment-free sequence comparison: measures, statistics and next-generation sequencing.". Briefings in bioinformatics 15: 343–353. doi:10.1093/bib/bbt067. PMID 24064230.
- ↑ Pinello, L; Lo Bosco, G; Yuan, GC (Nov 6, 2013). "Applications of alignment-free methods in epigenomics.". Briefings in bioinformatics 15: 419–430. doi:10.1093/bib/bbt078. PMID 24197932.
- ↑ La Rosa, M; Fiannaca, A; Rizzo, R; Urso, A (2013). "Alignment-free analysis of barcode sequences by means of compression-based methods.". BMC Bioinformatics. 14 Suppl 7: S4. doi:10.1186/1471-2105-14-S7-S4. PMID 23815444.
- ↑ Haubold, B (Nov 29, 2013). "Alignment-free phylogenetics and population genetics.". Briefings in bioinformatics 15: 407–418. doi:10.1093/bib/bbt083. PMID 24291823.
- ↑ 46.0 46.1 Domazet-Lošo, M; Haubold, B (Jun 1, 2011). "Alignment-free detection of local similarity among viral and bacterial genomes.". Bioinformatics (Oxford, England) 27 (11): 1466–72. doi:10.1093/bioinformatics/btr176. PMID 21471011.
- ↑ 47.0 47.1 Kolekar, P; Hake, N; Kale, M; Kulkarni-Kale, U (Dec 31, 2013). "WNV Typer: A server for genotyping of West Nile viruses using an alignment-free method based on a return time distribution.". Journal of Virological Methods 198C: 41–55. doi:10.1016/j.jviromet.2013.12.012. PMID 24388930.
- ↑ 48.0 48.1 Dimitrov, I; Naneva, L; Doytchinova, I; Bangov, I (Nov 7, 2013). "AllergenFP: allergenicity prediction by descriptor fingerprints.". Bioinformatics (Oxford, England) 30: 846–851. doi:10.1093/bioinformatics/btt619. PMID 24167156.
- ↑ 49.0 49.1 Gardner, SN; Hall, BG (Dec 9, 2013). "When Whole-Genome Alignments Just Won't Work: kSNP v2 Software for Alignment-Free SNP Discovery and Phylogenetics of Hundreds of Microbial Genomes.". PLoS ONE 8 (12): e81760. doi:10.1371/journal.pone.0081760. PMID 24349125.
- ↑ 50.0 50.1 Haubold, B; Krause, L; Horn, T; Pfaffelhuber, P (Dec 15, 2013). "An alignment-free test for recombination.". Bioinformatics (Oxford, England) 29 (24): 3121–7. doi:10.1093/bioinformatics/btt550. PMID 24064419.
- ↑ Xu, Z; Hao, B (Jul 2009). "CVTree update: a newly designed phylogenetic study platform using composition vectors and whole genomes.". Nucleic Acids Research 37 (Web Server issue): W174–8. doi:10.1093/nar/gkp278. PMC 2703908. PMID 19398429.
- ↑ Cheng, J; Cao, F; Liu, Z (May 2013). "AGP: a multimethods web server for alignment-free genome phylogeny.". Molecular Biology and Evolution 30 (5): 1032–7. doi:10.1093/molbev/mst021. PMID 23389766.
- ↑ Höhl, M; Rigoutsos, I; Ragan, MA (Feb 25, 2007). "Pattern-based phylogenetic distance estimation and tree reconstruction.". Evolutionary bioinformatics online 2: 359–75. PMID 19455227.
- ↑ Wang, Y; Liu, L; Chen, L; Chen, T; Sun, F (Jan 2, 2014). "Comparison of Metatranscriptomic Samples Based on k-Tuple Frequencies.". PLoS ONE 9 (1): e84348. doi:10.1371/journal.pone.0084348. PMID 24392128.