Aicardi syndrome
Not be confused with Aicardi-Goutières syndrome
| Aicardi syndrome | |
|---|---|
| Classification and external resources | |
| ICD-10 | Q04.0 |
| ICD-9 | 742.2 |
| OMIM | 304050 |
| DiseasesDB | 29761 |
| MedlinePlus | 001664 |
| eMedicine | ped/58 |
Aicardi syndrome is a rare genetic malformation syndrome characterized by the partial or complete absence of a key structure in the brain called the corpus callosum, the presence of retinal abnormalities, and seizures in the form of infantile spasms. Aicardi syndrome is theorized to be caused by a defect on the X chromosome as it has thus far only been observed in girls or in boys with Klinefelter syndrome. Confirmation of this theory awaits the discovery of the gene which causes Aicardi syndrome. Symptoms typically appear before a baby reaches about 5 months of age.
Signs and symptoms
Children are most commonly identified with Aicardi syndrome before the age of five months. A significant number of girls are products of normal births and seem to be developing normally until around the age of three months, when they begin to have infantile spasms. The onset of infantile spasms at this age is due to closure of the final neural synapses in the brain, a stage of normal brain development.
Genetics
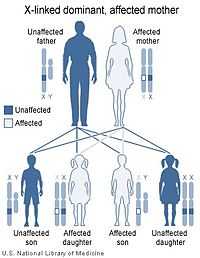 | 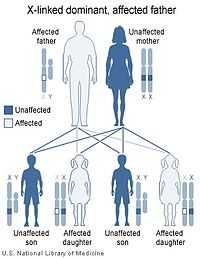 |
| X-linked dominant inheritance works differently depending upon whether the mother (left image) or father (right image) is the carrier of a gene that causes a disease or disorder. | |
Almost all reported cases of Aicardi syndrome have been in females. The few males that have been identified with Aicardi syndrome have proved to have 47 chromosomes including an XXY sex chromosome complement, a condition called Klinefelter syndrome.
Aicardi syndrome appears to be lethal in normal males who have only one X chromosome (and a Y chromosome). In other words, Aicardi syndrome appears to be inherited in an X-linked dominant pattern due to a mutant gene on the X chromosome that is lethal in XY males.
All cases of Aicardi syndrome are thought to be due to new mutations. No person with Aicardi syndrome is known to have transmitted the X-linked gene responsible for the syndrome to the next generation.
Diagnosis
Aicardi syndrome is typically characterized by the following triad of features - however, one of the "classic" features being missing does not preclude a diagnosis of Aicardi Syndrome, if other supporting features are present.[1]
- Partial or complete absence of the corpus callosum in the brain (agenesis of the corpus callosum);
- Eye abnormalities known as "lacunae" of the retina that are quite specific to this disorder; morning glory syndrome optic disc anomaly; and
- The development in infancy of seizures that are called infantile spasms.
Other types of defects of the brain such as microcephaly, polymicrogyria, porencephalic cysts and enlarged cerebral ventricles due to hydrocephalus are also common in Aicardi syndrome.
Treatment
Treatment of Aicardi syndrome primarily involves management of seizures and early/continuing intervention programs for developmental delays.
Additional complications sometimes seen with Aicardi syndrome include porencephalic cysts and hydrocephalus, and gastro-intestinal problems. Treatment for prencephalic cysts and/or hydrocephalus is often via a shunt or endoscopic fenestration of the cysts, though some require no treatment. Placement of a feeding tube, fundoplication, and surgeries to correct hernias or other gastrointestinal structural problems are sometimes used to treat gastro-intestinal issues.
Prognosis
The prognosis varies widely from case to case, depending on the severity of the symptoms. However, almost all people reported with Aicardi syndrome to date have experienced developmental delay of a significant degree, typically resulting in moderate to profound intellectual disability. The age range of the individuals reported with Aicardi syndrome is from birth to the mid 40s. There is no cure for this syndrome.
Epidemiology
Worldwide prevalence of Aicardi Syndrome is estimated at several thousand, with approximately 900 cases reported in the United States.[2]
History
This disorder was first recognized as a distinct syndrome in 1965 by Jean Aicardi, a French neurologist. A review article by Dr. Aicardi (Aicardi J, Aicardi syndrome: old and new findings, Int Pediatr. 1998;14(1):5-8) describes the syndrome.
References
External links
- GeneReviews/NCBI/NIH/UW entry on Aicardi Syndrome
- OMIM entries on Aicardi syndrome
- Phenotype and Management of Aicardi Syndrome: New Findings from a Survey of 69 Children
- Neurology India: Aicardi syndrome: A report of five Indian cases
- Aicardi Syndrome: Old and New Findings
- ninds.nih.gov
- UCSF Brain Development Research Program
- Baylor Department of Molecular and Human Genetics
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||