AMBER

AMBER (an acronym for Assisted Model Building with Energy Refinement) is a family of force fields for molecular dynamics of biomolecules originally developed by Peter Kollman's group at the University of California, San Francisco. AMBER is also the name for the molecular dynamics software package that simulates these force fields. It is maintained by an active collaboration between David Case at Rutgers University, Tom Cheatham at the University of Utah, Tom Darden at NIEHS, Ken Merz at Michigan State University, Carlos Simmerling at Stony Brook University, Ray Luo at UC Irvine, and Junmei Wang at Encysive Pharmaceuticals.
Force field
The term "AMBER force field" generally refers to the functional form used by the family of AMBER force fields. This form includes a number of parameters; each member of the family of AMBER force fields provides values for these parameters and has its own name.
Functional form
The functional form of the AMBER force field is[1]
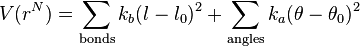
![+ \sum_\text{torsions} \sum_n \frac{1}{2} V_n [1+\cos(n \omega- \gamma)]](../I/m/74ec604269df35001d9b8c8491134705.png)
![+\sum_{j=1} ^{N-1} \sum_{i=j+1} ^N f_{ij}\biggl\{\epsilon_{ij}\biggl[\left(\frac{r_{0ij}}{r_{ij}} \right)^{12} - 2\left(\frac{r_{0ij}}{r_{ij}} \right)^{6} \biggr]+ \frac{q_iq_j}{4\pi \epsilon_0 r_{ij}}\biggr\}](../I/m/0d42465cf922c07d032a94894a6c240f.png)
Note that despite the term force field, this equation defines the potential energy of the system; the force is the derivative of this potential with respect to position.
The meanings of right hand side terms are:
- First term (summing over bonds): represents the energy between covalently bonded atoms. This harmonic (ideal spring) force is a good approximation near the equilibrium bond length, but becomes increasingly poor as atoms separate.
- Second term (summing over angles): represents the energy due to the geometry of electron orbitals involved in covalent bonding.
- Third term (summing over torsions): represents the energy for twisting a bond due to bond order (e.g. double bonds) and neighboring bonds or lone pairs of electrons. Note that a single bond may have more than one of these terms, such that the total torsional energy is expressed as a Fourier series.
- Fourth term (double summation over
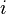 and
and 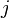 ): represents the non-bonded energy between all atom pairs, which can be decomposed into van der Waals (first term of summation) and electrostatic (second term of summation) energies.
): represents the non-bonded energy between all atom pairs, which can be decomposed into van der Waals (first term of summation) and electrostatic (second term of summation) energies.
The form of the van der Waals energy is calculated using the equilibrium distance (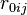 ) and well depth (
) and well depth ( ). The factor of
). The factor of 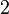 ensures that the equilibrium distance is . The energy is sometimes reformulated in terms of
ensures that the equilibrium distance is . The energy is sometimes reformulated in terms of 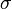 , where
, where 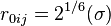 , as used e.g. in the implementation of the softcore potentials.
, as used e.g. in the implementation of the softcore potentials.
The form of the electrostatic energy used here assumes that the charges due to the protons and electrons in an atom can be represented by a single point charge (or in the case of parameter sets that employ lone pairs, a small number of point charges.)
Parameter sets
To use the AMBER force field, it is necessary to have values for the parameters of the force field (e.g. force constants, equilibrium bond lengths and angles, charges). A fairly large number of these parameter sets exist, and are described in detail in the AMBER software user manual. Each parameter set has a name, and provides parameters for certain types of molecules.
- Peptide, protein and nucleic acid parameters are provided by parameter sets with names beginning with "ff" and containing a two digit year number, for instance "ff99".
- GAFF (General AMBER force field) provides parameters for small organic molecules to facilitate simulations of drugs and small molecule ligands in conjunction with biomolecules.
- The GLYCAM force fields have been developed by Rob Woods for simulating carbohydrates.
Software
The AMBER software suite provides a set of programs for applying the AMBER forcefields to simulations of biomolecules. It is written in Fortran 90 and C with support for most major Unix-like systems and compilers. Development is conducted by a loose association of mostly academic labs. New versions are generally released in the spring of even numbered years; AMBER 10 was released in April 2008. The software is available under a site-license agreement, which includes full source, currently priced at US$400 for non-commercial and US$20,000 for commercial organizations.
Programs
- LEaP is used for preparing input files for the simulation programs
- Antechamber automates the process of parameterizing small organic molecules using GAFF
- SANDER (Simulated Annealing with NMR-Derived Energy Restraints) is the central simulation program and provides facilities for energy minimization and molecular dynamics with a wide variety of options
- pmemd is a somewhat more feature-limited reimplementation of sander by Bob Duke. It was designed with parallel processing in mind and has significantly better performance than sander when running on more than 8–16 processors
- pmemd.cuda has been made in order to run simulations on GPU enabled machines
- pmemd.amoeba was developed to handle the extra parameters in the polarizable AMOEBA force field.
- nmode calculates normal modes
- ptraj provides facilities for numerical analysis of simulation results. AMBER does not include visualization capabilities; visualization is commonly performed with VMD. Ptraj is now unsupported as of AmberTools 13.
- cpptraj is a rewritten version of ptraj made in C++ to provide faster analysis of simulation results. Several actions have been made parallelizable with OpenMP and MPI
- MM-PBSA allows for implicit solvent calculations on snap shots from molecular dynamics simulations
- NAB is a built in nucleic acid building environment made to aid in the process of manipulating proteins and nucleic acids where an atomic level of description will help with computation.
See also
References
- ↑ Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA (1995). "A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules". J. Am. Chem. Soc. 117: 5179–5197. doi:10.1021/ja00124a002.
Further reading
- Duan; Wu, Chun; Chowdhury, Shibasish; Lee, Mathew C.; Xiong, Guoming; Zhang, Wei; Yang, Rong; Cieplak, Piotr et al. (2003). "A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations". J Computational Chemistry 24 (16): 1999–2012. doi:10.1002/jcc.10349.