Vogt–Koyanagi–Harada syndrome
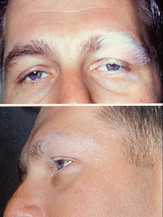
Vogt–Koyanagi–Harada syndrome (VKH syndrome) is an uncommon multisystem disease of presumed autoimmune etiology that is characterized by chronic, bilateral, diffuse, granulomatous uveitis with accompanying dermatologic, neurologic, and auditory involvement. VKH syndrome is named for ophthalmologists Alfred Vogt from Switzerland and Yoshizo Koyanagi and Einosuke Harada from Japan.[1][2][3]
Pathophysiology
VKH syndrome is an immune-mediated disease. The mechanism of the disease is thought to be T helper cell mediated autoimmune attack of melanocytes in the skin, uvea, central nervous system and inner ear. A new T-cell subset Th17 may play an important role in the initiation and maintenance of inflammatory disease when stimulated by the interleukin IL-23, thus producing IL-17.[4]
There is a higher rate of VKH syndrome in people of Asian, Latin, and Mediterranean descent
Clinical presentation

There are 4 stages of VKH syndrome: prodromal, acute uveitic, convalescent, and chronic recurrent.
- The prodromal stage is marked by flu-like symptoms. Patients present with headache, nausea, meningism, dysacusia (discomfort caused by loud noises or a distortion in the quality of the sounds being heard) and tinnitus.
- The acute uveitic stage is heralded by the onset of blurring of vision in both eyes and is marked by bilateral granulomatous anterior uveitis, variable degree of vitritis, thickening of the posterior choroid with elevation of the peripapillary retinal choroidal layer, hyperemia and edema of the optic nerve, and multiple serous retinal detachments.
- The convalescent stage occurs several weeks later and is marked by gradual depigmentation of the choroid, resulting in the classic orange-red discoloration, or sunset glow fundus appearance. Skin changes, including vitiligo, alopecia and poliosis typically appear during the this stage.
- The chronic recurrent stage is marked by repeated bouts of uveitis.
Diagnosis
The diagnosis of VKH syndrome is made by ophthalmologist or optometrist based on clinical presentation. Imaging techniques such as fluorescein angiography are used to confirm diagnosis.
Treatment
The acute stage of VKH syndrome is exquisitely responsive to early and aggressive treatment with topical, periocular and systemic corticosteroids and cycloplegic agents.
References
- ↑ A Vogt. Frühzeitiges Ergrauen der Zilien und Bemerkungen über den sogenannten plötzlichen Eintritt dieser Veränderung. Klinische Monatsblätter für Augenheilkunde, Stuttgart, 1906, 44: 228-242.
- ↑ Y. Koyanagi. Dysakusis, Alopecie und Poliosis bei schwerer Uveitis nicht traumatischen Ursprungs. Klinische Monatsblätter für Augenheilkunde, Stuttgart, 1929, 82: 194-211.
- ↑ E. Harada. Clinical study of nonsuppurative choroiditis. A report of acute diffuse choroiditis. Acta Societatis ophthalmologicae Japonicae, 1926, 30: 356.
- ↑ Bordaberry, MF (Nov 2010). "Vogt-Koyanagi-Harada disease: diagnosis and treatments update.". Current opinion in ophthalmology 21 (6): 430–5. doi:10.1097/ICU.0b013e32833eb78c. PMID 20829689.
Support
VKH Story http://vogtkoyanagiharadasyndrome.weebly.com/