Tajima's D
Tajima's D is a statistical test created by and named after the Japanese researcher Fumio Tajima.[1] The purpose of the test is to distinguish between a DNA sequence evolving randomly ("neutrally") and one evolving under a non-random process, including directional selection or balancing selection, demographic expansion or contraction, genetic hitchhiking, or introgression. A randomly evolving DNA sequence contains mutations with no effect on the fitness and survival of an organism. The randomly evolving mutations are called "neutral", while mutations under selection are "non-neutral". For example, you would expect to find that a mutation which causes prenatal death or severe disease to be under selection. When looking at the human population as a whole, we say that the population frequency of a neutral mutation fluctuates randomly (i.e. the percentage of people in the population with the mutation changes from one generation to the next, and this percentage is equally likely to go up or down) through genetic drift.
The strength of genetic drift depends on the population size. If a population is at a constant size with constant mutation rate, the population will reach an equilibrium of gene frequencies. This equilibrium has important properties, including the number of segregating sites 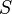 , and the number of nucleotide differences between pairs sampled (these are called pairwise differences). To standardize the pairwise differences, the mean or 'average' number of pairwise differences is used. This is simply the sum of the pairwise differences divided by the number of pairs, and is signified by
, and the number of nucleotide differences between pairs sampled (these are called pairwise differences). To standardize the pairwise differences, the mean or 'average' number of pairwise differences is used. This is simply the sum of the pairwise differences divided by the number of pairs, and is signified by  .
.
The purpose of Tajima's test is to identify sequences which do not fit the neutral theory model at equilibrium between mutation and genetic drift. In order to perform the test on a DNA sequence or gene, you need to sequence homologous DNA for at least 3 individuals. Tajima's statistic computes a standardized measure of the total number of segregating sites (these are DNA sites that are polymorphic) in the sampled DNA and the average number of mutations between pairs in the sample. The two quantities whose values are compared are both method of moments estimates of the population genetic parameter theta, and so are expected to equal the same value. If these two numbers only differ by as much as one could reasonably expect by chance, then the null hypothesis of neutrality cannot be rejected. Otherwise, the null hypothesis of neutrality is rejected.
A video explanation of Tajima's D, and its application to DNA sequences, is available online.
Scientific explanation
Under the neutral theory model, for a population at constant size at equilibrium:
![E[\pi ]=\theta =E\left[{\frac {S}{\sum _{{i=1}}^{{n-1}}{\frac {1}{i}}}}\right]=4N\mu](/2014-wikipedia_en_all_02_2014/I/media/4/d/5/d/4d5d2cd71a57d95be743265facced419.png)
for diploid DNA, and
![E[\pi ]=\theta =E\left[{\frac {S}{\sum _{{i=1}}^{{n-1}}{\frac {1}{i}}}}\right]=2N\mu](/2014-wikipedia_en_all_02_2014/I/media/9/0/1/2/9012ea4ec39e64ad3ca7e7a34390e6b2.png)
for haploid.
In the above formulas, S is the number of segregating sites, n is the number of samples, and i is the index of summation.
But selection, demographic fluctuations and other violations of the neutral model (including rate heterogeneity and introgression) will change the expected values of and , so that they are no longer expected to be equal. The difference in the expectations for these two variables (which can be positive or negative) is the crux of Tajima's D test statistic.
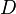 is calculated by taking the difference between the two estimates of the population genetics parameter
is calculated by taking the difference between the two estimates of the population genetics parameter  . This difference is called
. This difference is called 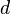 , and D is calculated by dividing by the square root of its variance
, and D is calculated by dividing by the square root of its variance 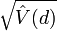 (its standard deviation, by definition).
(its standard deviation, by definition).
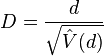
Fumio Tajima demonstrated by computer simulation that the statistic described above could be modeled using a beta distribution. If the value for a sample of sequences is outside the confidence interval then one can reject the null hypothesis of neutral mutation for the sequence in question.
Mathematical details
![D={\frac {d}{{\sqrt {{\hat {V}}(d)}}}}={\frac {{\hat {k}}-{\frac {S}{a_{1}}}}{{\sqrt {[e_{1}S+e_{2}S(S-1)]}}}}](/2014-wikipedia_en_all_02_2014/I/media/d/2/d/4/d2d4ac1ee24ad14750b62f6b5a4115de.png)
where

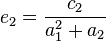
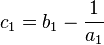
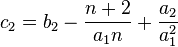
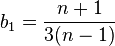
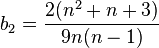
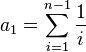
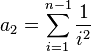
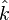 and
and 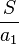 are two estimates of the expected number of single nucleotide polymorphisms (SNPs)between two DNA sequences under the neutral mutation model in a sample size
are two estimates of the expected number of single nucleotide polymorphisms (SNPs)between two DNA sequences under the neutral mutation model in a sample size 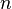 from an effective population size
from an effective population size 
The first estimate is the average number of SNPs found in (n choose 2) pairwise comparisons of sequences 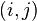 in the sample
in the sample
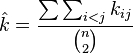
The second estimate is derived from the expected value of  , the total number of polymorphisms in the sample
, the total number of polymorphisms in the sample
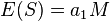
Tajima defines  , whereas Hartl & Clark use a different symbol to define the same parameter
, whereas Hartl & Clark use a different symbol to define the same parameter  .
.
Historical example
The genetic mutation which causes sickle-cell anemia is non-neutral because it affects survival and fitness. People homozygous for the mutation have the sickle-cell disease, while those without the mutation (homozygous for the wild-type allele) do not have the disease. People with one copy of the mutated allele (heterozygous) do not have the disease, but instead are resistant to malaria. Thus in Africa, where there is a prevalence of the malaria parasite Plasmodium falciparum that is transmitted through mosquitos Anopheles, there is a selective advantage for heterozygous individuals. Meanwhile, in countries such as the USA where the risk of malaria infection is low, the population frequency of the mutation is lower.
Example
Suppose you are a geneticist studying an unknown gene. As part of your research you get DNA samples from four random people (plus yourself). For simplicity, you label your sequence as a string of zeroes, and for the other four people you put a zero when their DNA is the same as yours and a one when it is different. (For this example, the specific type of difference is not important.)
1 2 Position 12345 67890 12345 67890 Person Y 00000 00000 00000 00000 Person A 00100 00000 00100 00010 Person B 00000 00000 00100 00010 Person C 00000 01000 00000 00010 Person D 00000 01000 00100 00010
Notice the four polymorphic sites (positions where someone differs from you, at 3, 7, 13 and 19 above). Now compare each pair of sequences and get the average number of polymorphisms between two sequences. There are "five choose two" (ten) comparisons that need to be done.
Person Y is you!
You vs A: 3 polymorphismsPerson Y 00000 00000 00000 00000 Person A 00100 00000 00100 00010You vs B: 2 polymorphismsPerson Y 00000 00000 00000 00000 Person B 00000 00000 00100 00010You vs C: 2 polymorphismsPerson Y 00000 00000 00000 00000 Person C 00000 01000 00000 00010You vs D: 3 polymorphismsPerson Y 00000 00000 00000 00000 Person D 00000 01000 00100 00010A vs B: 1 polymorphismPerson A 00100 00000 00100 00010 Person B 00000 00000 00100 00010A vs C: 3 polymorphismsPerson A 00100 00000 00100 00010 Person C 00000 01000 00000 00010A vs D: 2 polymorphismsPerson A 00100 00000 00100 00010 Person D 00000 01000 00100 00010B vs C: 2 polymorphismsPerson B 00000 00000 00100 00010 Person C 00000 01000 00000 00010B vs D: 1 polymorphismPerson B 00000 00000 00100 00010 Person D 00000 01000 00100 00010C vs D: 1 polymorphismPerson C 00000 01000 00000 00010 Person D 00000 01000 00100 00010
The average number of polymorphisms is 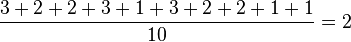 .
.
The lower-case d described above is the difference between these two numbers—the average number of polymorphisms found in pairwise comparison (2) and the total number of polymorphic sites (4). Thus 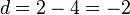 .
.
Since this is a statistical test, you need to assess the significance of this value. A discussion of how to do this is provided below.
Determining significance
When performing a statistical test such as Tajima's D, the critical question is whether the value calculated for the statistic is unexpected under a null process. For Tajima's D, the magnitude of the statistic is expected to increase the more the data deviates from a pattern expected under a population evolving according to the standard coalescent model.
A negative Tajima's D signifies an excess of low frequency polymorphisms relative to expectation, indicating population size expansion (e.g., after a bottleneck or a selective sweep) and/or purifying selection. A positive Tajima's D signifies low levels of both low and high frequency polymorphisms, indicating a decrease in population size and/or balancing selection. However, calculating a conventional "p-value" associated with any Tajima's D value that is obtained from a sample is impossible. Briefly, this is because there is no way to describe the distribution of the statistic that is independent of the true, and unknown, theta parameter (no pivot quantity exists). To circumvent this issue, several options have been proposed.
Tajima (1989) found an empirical similarity between the distribution of the test statistic and a beta distribution with mean zero and variance one. He estimated theta by taking Watterson's estimator and dividing it the number of samples. Simulations have shown this distribution to be conservative,[2] and now that the computing power is more readily available this approximation is not frequently used.
A more nuanced approach was presented in a paper by Simonsen et al.[3] These authors advocated constructing a confidence interval for the true theta value, and then performing a grid search over this interval to obtain the critical values at which the statistic is significant below a particular alpha value. An alternative approach is for the investigator to perform the grid search over the values of theta which they believe to be plausible based on their knowledge of the organism under study. Bayesian approaches are a natural extension of this method.
A very rough rule of thumb to significance is that values greater than +2 or less than -2 are likely to be significant. This rule is based on an appeal to asymptotic properties of some statistics, and thus +/- 2 does not actually represent a critical value for a significance test.
Finally, genome wide scan's of Tajima's D in sliding windows along a chromosomal segment are often performed. With this approach, those regions that have a value of D that greatly deviates from the bulk of the empirical distribution of all such windows are reported as significant. This method does not assess significance in the traditional statistical sense, but is quite powerful given a large genomic region, and is unlikely to falsely identify interesting regions of a chromosome if only the greatest outliers are reported.
References
- ↑ Tajima, F. (Nov 1989). "Statistical method for testing the neutral mutation hypothesis by DNA polymorphism.". Genetics 123 (3): 585–95. PMC 1203831. PMID 2513255.
- ↑ Fu, YX.; Li, WH. (Mar 1993). "Statistical tests of neutrality of mutations.". Genetics 133 (3): 693–709. PMC 1205353. PMID 8454210.
- ↑ Simonsen, KL.; Churchill, GA.; Aquadro, CF. (Sep 1995). "Properties of statistical tests of neutrality for DNA polymorphism data.". Genetics 141 (1): 413–29. PMC 1206737. PMID 8536987.
- Notes
- Principles of Population Genetics, 4th ed. Daniel L. Hartl & Andrew G. Clark. Sinauer Associates, Inc. 2007
Computational tools for Tajima's D test
- DNAsp (Windows)
- Variscan (Mac OS X, Linux, Windows)
- Arlequin (Windows)
- Online view of Tajima's D values in human genome
- Online computation of Tajima's D
- MEGA4 or MEGA5
- Bio::PopGen::Statistics in BioPerl
| |||||||||||